Nat Rev Immunol :肿瘤免疫治疗中的细胞因子释放综合征和相关神经毒性
时间:2022-10-27 05:59:58 热度:37.1℃ 作者:网络
摘要
最近在肿瘤治疗领域发生了范式转换(paradigm shift),基于 T 细胞工程的细胞免疫治疗已加入传统的抗癌药(如化疗、放疗和靶向特定信号通路的小分子药物)。在肿瘤免疫学、基因工程和细胞生产的科学发展基础上,新型患者特异性细胞疗法得以迅速采用,以 CD19 表达恶性肿瘤为靶点的嵌合抗原受体 (CAR)T 细胞疗法的治愈潜力便可充分说明这一点。然而许多患者的临床获益可能是有代价的,高达1/3的患者会发生与诱导强效免疫效应应答直接相关的显著毒性,其中最常见的免疫介导毒性是细胞因子释放综合征和免疫效应细胞相关的神经毒性综合征。本综述讨论了我们目前对其病理生理学和临床特征的理解,以及开发用于其预防和/或管理的新型治疗药物。
过继转移的肿瘤抗原特异性 T 细胞经基因工程改造后可表达嵌合抗原受体 (CAR),在部分类型的肿瘤患者中获得了持久的临床缓解,特别是表达 CD19 的难治性和复发性 B 细胞恶性肿瘤。CAR 是合成的抗原识别受体,包括抗体衍生的单链可变片段 (scFv)、铰链和跨膜结构域及胞内信号结构域。胞内信号域一般为CD3ζ信号域和共刺激信号域,如CD28和4-1BB(也称为TNFRSF9),胞内信号域及工程 T 细胞的细胞表型一旦被表达抗原的靶细胞激活,就会影响 CAR T 细胞的特异性细胞因子分泌和体内增殖能力。
尽管在临床上取得了成功,但CAR T细胞也会导致显著毒性,这与其诱导的强大的免疫效应反应直接相关。CAR T 细胞生成的两种主要毒性在早期小鼠模型中均未显示,但最终在临床试验中却发生,即细胞因子释放综合征 (CRS) 和免疫效应细胞相关神经毒性综合征(ICANS;通常称为神经毒性)。CRS 通常以发热和全身症状(如寒战、不适和厌食)为起始,发热可为高级别并持续数天。在重度病例中CRS表现为全身炎症反应的其他特征,包括低血压、缺氧和/或器官功能障碍,器官功能障碍可能继发于低血压或缺氧,但也可由细胞因子释放的直接作用所导致。CRS患者可发生所有主要器官系统的功能障碍,包括心脏、肺、肝、肾和胃肠道系统等,但如果及时识别和管理 CRS 的症状和体征,多数患者的器官功能障碍是可预防或可逆的。
ICANS 通常表现为中毒性脑病,以找词困难、意识模糊、言语困难、失语、精细运动技能受损和嗜睡等为起始,严重患者可发生癫痫发作、运动无力、脑水肿和昏迷。大多数出现 ICANS 临床特征的患者既往发生过CRS,因此 CRS 被认为是ICANS的‘始发事件’或辅因子。ICANS多发生于 CRS 症状消退后(图1),虽然频率较低,但CRS 和ICANS也可同时出现。此外与 CRS 相似,ICANS在多数患者中是可逆的,无永久性神经功能缺损。
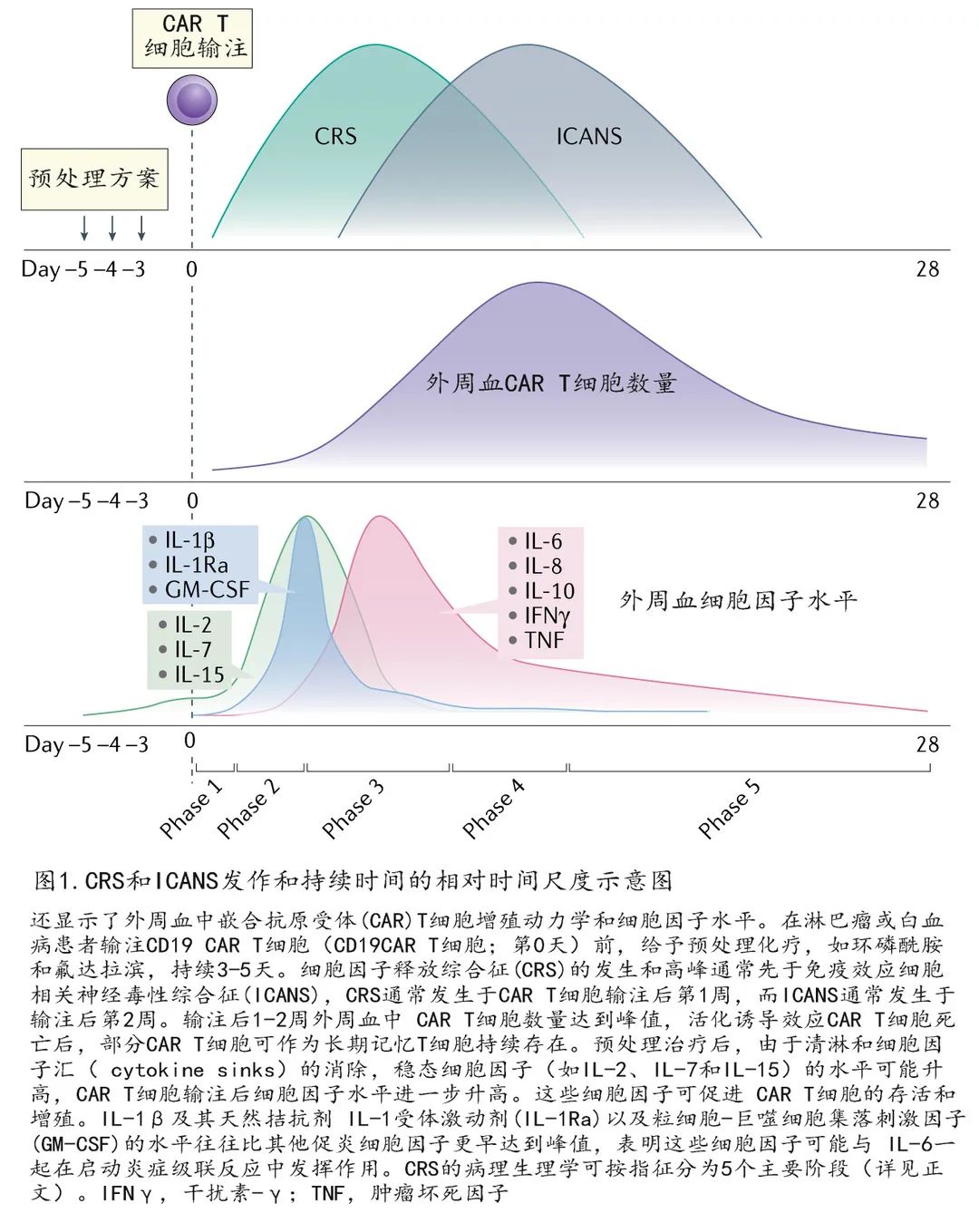 目前CAR T细胞的大多数临床应用是针对高危恶性疾病,在血液肿瘤学领域内多为靶向CD19的 CAR T 细胞(CD19CAR T细胞),因此这也是本综述的重点。但应注意的是,CAR T多种临床适应症中均观察到了与本文所述相似的毒性,具有广泛的 CAR T 细胞特异性。用于自身免疫性疾病和实体器官移植耐受等非恶性适应症的基因修饰免疫细胞的开发正取得突飞猛进的进展,其风险-获益平衡并不利于潜在毒性,需要改善毒性管理或降低免疫治疗的毒性,因此更好地了解 CRS 和 ICANS 的临床特征和病理生理机制具有重要意义。
目前CAR T细胞的大多数临床应用是针对高危恶性疾病,在血液肿瘤学领域内多为靶向CD19的 CAR T 细胞(CD19CAR T细胞),因此这也是本综述的重点。但应注意的是,CAR T多种临床适应症中均观察到了与本文所述相似的毒性,具有广泛的 CAR T 细胞特异性。用于自身免疫性疾病和实体器官移植耐受等非恶性适应症的基因修饰免疫细胞的开发正取得突飞猛进的进展,其风险-获益平衡并不利于潜在毒性,需要改善毒性管理或降低免疫治疗的毒性,因此更好地了解 CRS 和 ICANS 的临床特征和病理生理机制具有重要意义。
在此我们讨论了迄今为止的已知数据,并阐述了其如何影响预防和/或管理这些毒性的新型治疗药物的开发。此外对 CRS 和 ICANS 的深入了解也与其他全身性细胞因子介导的炎症性疾病有更广泛的意义,如脓毒性休克、巨噬细胞活化综合征(BOX 1)、神经炎症性疾病和新出现的病原体感染,如SARS-CoV-2(BOX 2),以及其他治疗干预措施,如 IL-1 阻断在肿瘤免疫治疗中的潜在作用(BOX 3)。如下文所述,我们对 CAR T 细胞治疗相关 CRS 和 ICANS 病理生理学的理解始于临床终于实验室研究,现在通过动物模型的详尽研究又得以进一步扩展。



CRS的病理生理学
CRS 的病理生理学可分为5个主要阶段。第1阶段为 CAR T 细胞输注到患者体内后转移到肿瘤部位,CAR介导识别表达抗原的靶细胞。第2阶段,肿瘤部位出现 CAR T 细胞增殖,活化的 CAR T 细胞和肿瘤微环境的细胞组分可原位生成细胞因子,激活“旁观者”内源性免疫细胞,直接和间接杀死肿瘤细胞并发生CRS。第3阶段,外周血中的细胞因子升高及CAR T细胞群扩增,与全身炎症反应相关,可导致多个组织和器官的内皮损伤和血管渗漏及其相关效应,包括缺氧、低血压和/或器官损伤。第4阶段,细胞因子扩散和 CAR T 细胞移行,内源性 T 细胞和外周活化单核细胞进入脑脊液 (CSF) 和中枢神经系统 (CNS) ,包括血脑屏障 (BBB) 破坏,这与 ICANS 发作一致。第5阶段,活化诱导的 T 细胞死亡及肿瘤根除导致血清细胞因子降低和全身炎症反应减少,CRS和/或 ICANS 症状结束,并可能持续存在长期记忆 CAR T 细胞。CRS 和 ICANS 发作和持续时间的相对时间如图1所示。
CD19CAR T 细胞的临床前研究并未发生CRS,仅在启动 CAR T 细胞的 I 期临床试验后才发现。尽管患者出现的发热和低血压与血清细胞因子即刻升高相关,但这种偶发结局(高肿瘤负荷患者中更常见)的潜在机制最初尚不清楚。现已清楚几乎所有接受 CD19CAR T 细胞治疗的患者都会出现一定程度的CRS;在注册试验中,高达1/3的 B 细胞急性淋巴细胞白血病患者出现重度CRS,高达一半的患者出现ICANS,但CRS 和 ICANS 的实际发生率可能低于此。不同阻断抗体的实证检验很快确定 IL-6 是 CRS 的关键介质,因此托珠单抗(一种通过 IL-6 受体 (IL-6R) 阻断信号传导的单抗)成为 CRS 管理的主要药物。由于活化 T 细胞可生成IL-6,因此最初假定 CAR T 细胞本身是其主要来源,尽管有观察结果提示了额外的可能促成因素,然而随后的研究确定巨噬细胞和单核细胞谱系细胞是 IL-6 的来源。值得注意的是,尽管一些研究中 CAR T 细胞(包括 CD28 或 4-1BB 信号结构域)的活化动力学和细胞因子生成能力之间似乎存在显著差异,但与两种CAR 相关的 CRS 均高度相似。尽管CD28 CAR T 细胞诱导的 CRS 倾向于早于4-1BB CAR T 细胞,但患者血清中的细胞因子谱在细胞因子和趋化因子的峰值水平方面几乎没有差异,这与其共同的病理生理机制相一致。IL-6、IL-8、IL-1受体拮抗剂 (IL-1Ra)、CC-趋化因子配体 2(CCL2) 和CCL3(并非主要的 T 细胞衍生产物)通常升高,这表明 CRS 可能存在一种超出 CAR T 细胞本身并涉及宿主细胞的共同机制。CRS 的病理生理学示意图见图2。

CRS 与系统性炎症反应综合征如噬血细胞性淋巴组织细胞增生症和巨噬细胞活化综合征的相似之处在BOX 1中讨论。尽管观察认为这些和其他迟发型炎症毒性与 CAR T 细胞特异性无关,但数据表明CD22CAR T细胞(开发用作靶向继发于 CD19 抗原逃逸后复发的肿瘤)发生频率更高。
细胞相互作用和分子学介质。两种小鼠模型最终表明,CRS是由 CAR T 细胞和宿主细胞参与的多细胞网络所导致,其中巨噬细胞和单核细胞系细胞最重要。一项研究表明,在CD19+淋巴瘤异种移植模型中,肿瘤部位的多种内源性细胞群(包括树突状细胞、单核细胞和巨噬细胞)可生成IL-6,且巨噬细胞远超其他细胞类型。在这种异基因情况下,CAR T 细胞生成的小鼠 IL-6 水平大大超过人 IL-6 。远端无肿瘤部位未诱导 IL-6 生成,从而支持 CRS 起源于局部但产生全身病理学的观点。另一项研究表明,使用患者来源的白血病细胞系(用氯膦酸盐耗尽巨噬细胞)的人源化NGS小鼠,或在治疗性 CAR T 细胞给药前给予CAR T 细胞介导的靶向作用,可消除 IL-6 生成和显著CRS。CRS 期间分离的白细胞单细胞 RNA 测序数据证实,单核细胞系细胞是 IL-6 的起源。在两种模型中,以及根据临床经验,阻断 IL-6 可较大程度减轻 CRS 相关毒性。两项研究均进一步揭示了IL-1(一种单核和巨噬细胞来源的细胞因子)也是 CRS 相关毒性的强效驱动因子。
CRS 期间巨噬细胞募集或活化的潜在触发因素正浮现出来。CAR T 细胞本身必须被激活才能诱导髓系细胞生成细胞因子,这与临床观察结果一致,即 CAR T 细胞治疗无效的患者 CRS 为轻度甚至无。目前尚不确定 CAR T 细胞和宿主骨髓细胞之间是否需要接触依赖性相互作用,CD40-CD40L 相互作用虽然不是引起 CRS 所必需的,但可加剧巨噬细胞活化,从而增加 IL-6 的生成并加重CRS;其他哪些接触依赖性机制(如果有)可能对 CRS 的发展或扩增至关重要,则仍有待确定。T 细胞表面分子(如整合素 LFA1 和共刺激分子 CD28) 可与骨髓细胞强烈表达的同源受体(分别为 ICAM1 和 CD80 或CD86)相互作用,基于CD28-CD80/CD86轴双向信号转导和诱导骨髓细胞生成 IL-6 的潜在作用,其可能在CRS 中特别有研究价值,研究也证实阻断这些相互作用可能降低 CRS 的严重程度。
细胞因子介质:IL-6和IL-1。如上所述,细胞因子 IL-6 和 IL-1 的非接触依赖性相互作用在 CRS 的病理生理学中具有重要作用。IL-6 是一种多效性细胞因子,同时具有促炎和抗炎作用,它主要由巨噬细胞和髓系的其他细胞产生,以自分泌的方式发挥作用,结合其他炎症信号促进巨噬细胞成熟和活化。IL-6R 主要由免疫细胞(包括小胶质细胞)和肝细胞表达。IL-6 可以通过顺式信号和反式信号传导传递信号和促进炎症,可溶性IL-6–IL-6R与广泛表达的膜结合 gp130 形成复合物时,反式信号在免疫系统外具有广泛的作用。例如,IL-6除了控制急性期反应外,还参与体温过高、糖代谢、神经内分泌系统和食欲的调节,但IL-6在 CRS 中的确切作用仍不明确。阻断IL-6 可导致许多患者大多数症状逆转和全面的细胞因子下调,临床前模型还发现 IL-6 可调节 CRS 死亡率,以及通过诱导一氧化氮合酶 (iNOS) 和生成一氧化氮 (NO) 来促进巨噬细胞活化。
IL-1 也是一种具有多种功能的多效性细胞因子,主要由单核细胞和巨噬细胞生成。IL-1 受体 (IL-1R1) 广泛表达,负责转导促炎信号转导。IL-1 可诱导组织产生下游促炎细胞因子(如IL-6)以及一系列可组织成熟细胞和募集免疫细胞的趋化因子,还可活化促炎脂质介质(如前列腺素E2,可促进水肿)的生成,诱导急性期蛋白并向下丘脑发出信号诱导发热,以及向垂体和肾上腺发出信号,对循环系统具有直接和间接作用。
两项独立研究均发现 IL-1 是 CRS 的关键介质(图2):在人源化异种移植模型中,用 IL-1R 拮抗剂阿那白滞素阻断 IL-1 诱导的信号转导可保护小鼠免免于体重减轻和发热,并预防 CRS 相关死亡;在一个SCID–beige异种移植模型中,阿那白滞素可保护小鼠免受 CRS 相关的死亡,并减少巨噬细胞表达 iNOS;经改造表达 IL-1Ra 的 CAR T 细胞同样可提供对 CRS 致死性的保护作用。重要的是,在上述两种小鼠模型中,预防性启动 IL-1 抑制时,CAR T细胞的抗肿瘤疗效仍保持完整并同时抑制CRS。抗 IL-1 治疗的治疗时间可能对其疗效至关重要。有趣的是在人源化 NSG 小鼠模型中,与阻断 IL-1R 相比,阻断IL-6未改善巨噬细胞对大脑的浸润。
基于动物研究,目前已经启动了多项临床试验评估阿那白滞素在 CRS 和神经毒性预防中的作用(ClinicalTrials.gov NCT04148430、NCT04205838、NCT03430011、NCT04432506和NCT04359784)。鉴于IL-1β阻断药物(如单抗canakinumab)的临床可及性,未来的临床前研究应更多地阐明IL-1α与IL-1β在 CRS 级联反应中的具体作用,因为这些细胞因子在促炎功能中显示出较大的重叠,但也可通过表达和疾病背景的差异所区分。
损伤相关分子模式和其他可溶性介质。CAR T 细胞倾向于通过焦亡的炎症过程而非凋亡来诱导细胞死亡,从而导致损伤相关分子模式(如 ATP 和HMGB1)释放。肿瘤细胞释放损伤相关分子模式可导致体外巨噬细胞活化,生成 IL-6 和IL-1,而在SCID–beige异种移植模型中抑制体内焦亡可降低 CRS 相关死亡率(但也可能影响 CAR T 细胞的细胞溶解活性)。
粒细胞-巨噬细胞集落刺激因子 (GM-CSF) 由包括活化的 CAR T 细胞在内的多种细胞所生成,阻断其可在体外消除单核细胞生成 IL-6 和其他细胞因子。然而IL-6的生成极其强大,在异种移植模型中CRS最终是致命的,人 T 细胞来源的 GM-CSF 未与同源小鼠受体发生交叉反应,但GM-CSF可能仍是 CRS 的一个促进因素而并不需要CRS。在人 PBMC 异种移植模型中,阻断小鼠和人 GM-CSF 均可降低 IL-6 生成(图2、3)。
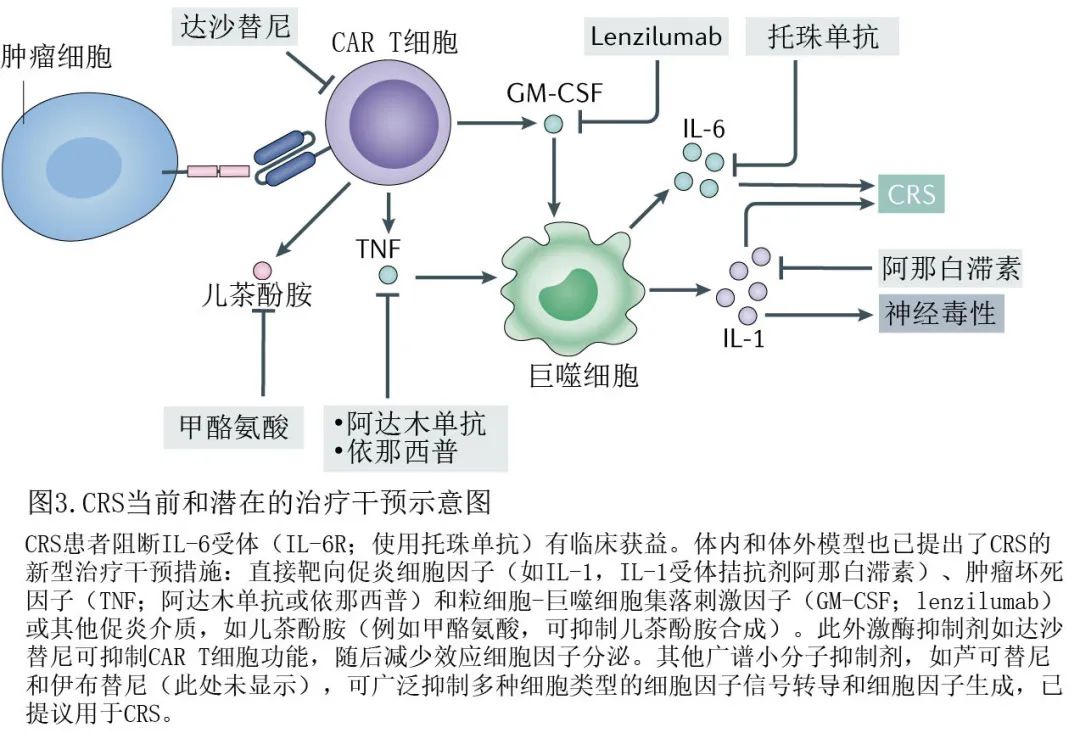
肿瘤坏死因子 (ΤΝF) 是另一种多效性细胞因子,可通过激活 T 细胞、巨噬细胞、单核细胞和免疫系统的其他细胞介导炎症,而与 IL-1 和 IL-6 不同的是TNF也有直接杀肿瘤活性。最近有学者在HER2 人源化小鼠乳腺肿瘤病毒 (MMTV) 乳腺癌模型中提出,TNF 是 CD3 和 HER2 双特异性抗体诱导 CRS 的潜在因素。在该模型中,TNF抑制剂预处理可降低循环 IL-6 水平(某些情况下可降低 IL-1 水平)而未损害抗肿瘤疗效。在SCID–beige异种移植模型中,阻断TNF可显著减少骨髓细胞生成IL-6及消除了 CRS 相关死亡率,但也会降低 CAR T 细胞的抗肿瘤活性,当然也要取决于 CAR 结构(未发表数据)(图2,3)。
干扰素-γ (IFNγ) 由活化T 细胞大量生成,可通过 iNOS 诱导促进巨噬细胞成熟增强 NO 生成,还可通过松开紧密连接(loosening tight junctions)促进包括血脑屏障在内的其他组织的通透性增加。重度 CRS 患者中IFNγ的血清浓度可增加高达100倍,在巨噬细胞活化综合征中也存在此现象(BOX 1),尽管有这一观察结果,但IFNγ在 CRS 中的特异性致病作用目前尚无报道。
肾上腺系统也涉及 CRS 的发生和维持,因为儿茶酚胺肾上腺素和去甲肾上腺素会直接影响 CAR T 细胞活化和随后的细胞因子释放。给予心房利钠肽(ANP,电解质和细胞外液量的调节剂和细胞因子分泌抑制剂)或通过甲酪氨酸抑制儿茶酚胺合成可减少 CAR T 细胞在体外和体内生成的细胞因子,在异种移植小鼠模型中还可降低死亡率(图2、3)。在 B 细胞急性淋巴细胞白血病的同源小鼠模型中,低剂量甲基酪氨酸不会损害抗肿瘤疗效。有趣的是,免疫细胞不仅可被儿茶酚胺激活,而且激活后可天然生产儿茶酚胺,从而创建了一个正反馈回路。然而ANP 失调可促进水肿、血压降低和电解质失衡,它们在 CRS 病理学均常见。因此阻断儿茶酚胺受体安全且可行,尤其是在 CRS 发作时,这将引起关注。
ICANS的病理生理学
尽管 ICANS 的临床特征很容易识别,但其病理生理学仍知之甚少。最近的动物模型表明,除了各种促炎性细胞因子的作用外,内皮细胞活化和血脑屏障破坏还可直接导致神经元细胞损伤,但这些 CAR T 细胞介导神经毒性的模型受到缺乏人细胞因子和造血细胞以及伴随异种移植抗宿主病发生的限制。尽管如此,最近在小鼠和非人灵长类动物中的研究还是产生了重要的见解,它们与输注 CAR T 细胞后发生 ICANS 的临床试验数据一起,提高了我们对 ICANS 病理生理学的理解(图4)。
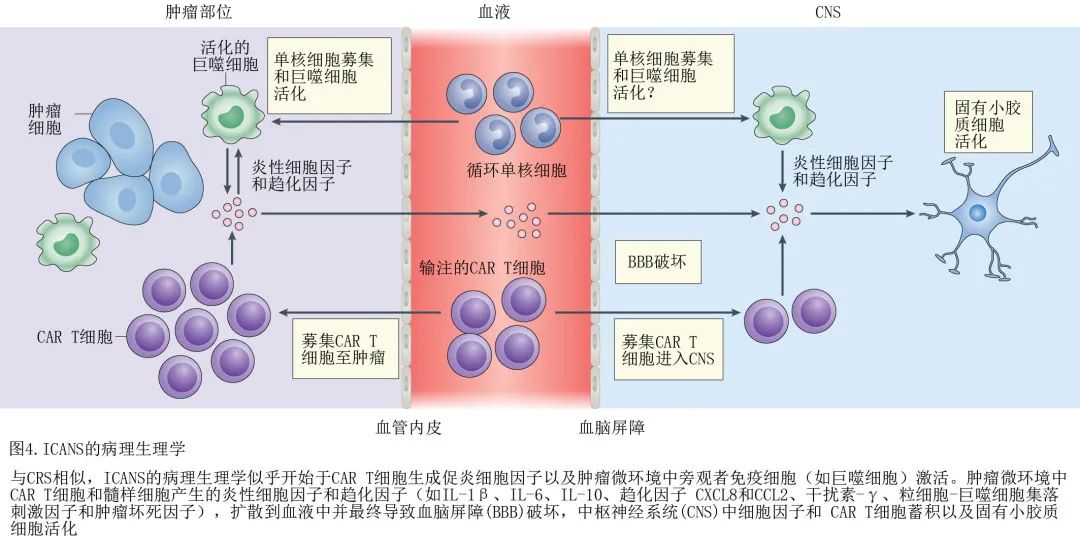
血管通透性、内皮破坏和神经胶质细胞损伤。ICANS 患者 CSF 中蛋白、CD4+ T细胞、CD8+ T细胞和 CAR T 细胞水平升高,表明血脑屏障完整性丧失;而临床研究显示 CAR T 细胞数量和 CSF 中细胞因子水平均与 ICANS 严重程度相关。
患者中观察到的重度 CRS 和 ICANS 的部分生化特征(如低纤维蛋白原血症和纤维蛋白降解产物增加)与弥散性血管内凝血和内皮细胞破坏的特征相同,后者通常见于脓毒症和与血管通透性增加相关的危重疾病。事实上确实有证据表明重度 ICANS 患者存在血管渗漏。在一项133例接受 CD19CAR T 细胞治疗的患者的单中心研究中,便描述了健康调节内皮细胞活化的血管生成素 (ANG)-TIE2 轴的变化。血小板和血管周围细胞生成ANG1,当与其内皮受体 TIE2 结合时,可稳定内皮。炎性细胞因子激活内皮细胞后,ANG2从内皮Weibel–Palade小体中释放并取代ANG1,进一步增加内皮细胞活化和微血管通透性。与此机制一致的是,与神经毒性较轻的患者相比,重度 ICANS 患者血清 ANG2 与 ANG1 的比值统计学显著增加,同时伴血管性血友病因子 (vWF) 和 CXC-趋化因子配体8(CXCL8) 浓度升高,它们均由血小板和血管周围细胞产生。此外,清淋和 CAR T 细胞给药前血清 ANG2 与 ANG1 比值升高的患者发生 ICANS 的风险较高。进一步的数据指出,在严重 ICANS 患者中扣留(sequestrate)高分子量 vWF 多聚体会导致凝血病。然而控制基线和干扰 ANG2 和 ANG1 水平的因素仍不明确。
除 BBB 破坏和血管通透性增加外,在 CD19CAR T 细胞治疗后发生 ICANS 的儿童和年轻成人患者中还报告了神经胶质细胞损伤。在一项43例患者的队列中,急性神经毒性患者CSF 中 GFAP 和 S100b 水平显著升高。GFAP 是星形胶质细胞损伤的一个经过充分验证的标记物(无论原因如何),而 CSF 中的 S100b 是星形胶质细胞活化的标记物。此外这项研究发现,CSF中IL-6、IL-10、IFNγ和颗粒酶 B 水平升高也与神经毒性相关。
细胞因子及其细胞来源。在人源化 NSG 小鼠模型中,输注携带 CD28 或 4-1BB 共刺激结构域的人 CD19CAR T 细胞会导致 B 细胞再生障碍性贫血、CRS和神经毒性。这些小鼠在初始 CRS 后出现延迟致死性ICANS,在部分患者中也观察到该情况(通常< 1%)。然而在该模型中,通过 IL-6R 阻断信号转导对神经毒性无影响;相比之下,阻断IL-1R可消除 CRS 和神经毒性而不影响 CAR T 细胞的功效。单核细胞消融对 CAR T 细胞增殖和群体扩增有负面影响。这一差异可能是由于 IL-1R 拮抗剂阿那白滞素可穿过BBB,而无明确证据表明 IL-6R 特异性单抗托珠单抗可穿透CNS。
利用免疫功能正常的恒河猴建立的非人灵长类动物ICANS模型最近报道,这些灵长类动物在输注携带 4-1BB 共刺激结构域的 CD20CAR T 细胞后出现了与 ICANS 一致的特征。尽管在神经毒性达峰期间观察到 CSF 和脑实质中的 CAR T 细胞和非 CAR T 细胞数量均增加,但 CAR T 细胞的数量高于非 CAR T 细胞。这与高浓度的IL-6、CXCL8、IL-1Rα、CXCL9、CXCL11、GM-CSF和血管内皮生长因子有关,它们在CSF 中的水平高于相应的血清样本。CAR T 细胞输注后 8d 可见不同程度的组织学全脑炎,包括多灶性脑膜炎和血管周围 T 细胞浸润,与外周血中 CAR T 细胞增殖峰值水平相吻合。详细的表型分析发现,与非 CAR T 细胞相比,CAR T细胞表面整合素 VLA4 的表达显著增加,可能促进 CAR T 细胞向 CNS 的转运增加。综上所述,这些发现表明,驱动 ICANS 的机制可能包括促炎性细胞因子和 CAR T 细胞在 CNS 中的积累,但它们的相对贡献迄今尚不清楚。
大量临床试验表明,血清中多种细胞因子水平升高与发生 ICANS 的风险相关。在多项研究(使用多种 CAR 结构和不同的靶恶性肿瘤)中均发现,患者中持续增加的细胞因子包括IL-2、IL-6、IL-10和IL-15。然而,即使小鼠模型已表明受体单核细胞来源的免疫细胞在细胞因子分泌以及 CRS 和 ICANS 发病机制中具有明确作用,但在临床试验中无法确定 ICANS 患者中细胞因子的特定细胞来源。但在发生重度 ICANS 的患者 CSF 中观察到骨髓细胞数量显著增加。
CNS 中的靶抗原表达。临床试验数据表明,发生ICANS不需要 CNS 中存在抗原阳性肿瘤细胞。而且值得注意的是,当 CAR T 细胞在多形性胶质母细胞瘤患者鞘内或瘤内输注时,这些患者并不会发生ICANS。然而最近的一项研究中,单细胞 RNA 测序分析证明了 CD19 在人脑壁细胞(包括周细胞和血管平滑肌细胞)中表达,因此脱靶(on-target off-tumour)效应可能导致与 CD19CAR T 细胞神经毒性相关。该观察结果可以解释,为什么与靶向CD20、CD22和BCMA(也称为TNFRSF17)的治疗相比,CD19靶向治疗的 ICANS 发生率更高,但也可能有替代或其他机制。在 CD22CAR T 细胞治疗时,人脑小胶质细胞表达的 CD22 与 ICANS 的较高发生率或严重程度均无关。小胶质细胞是一种特殊的吞噬细胞,在 CNS 中可持续存在数十年;它们吞噬髓鞘碎片和蛋白质聚集体,从而防止神经元细胞损伤,维护大脑稳态和功能。利用CRISPR–Cas9敲除筛选并结合 RNA 测序,已经确定了 CD22 作为小胶质细胞吞噬作用的负调控因子的作用(之前未曾报道)。在老年小胶质细胞小鼠中CD22上调,从而损害体内髓鞘碎片、β-淀粉样低聚物和α-突触核蛋白原纤维的清除,而给予 CD22 阻断抗体可逆转小胶质细胞功能障碍,改善吞噬作用和认知功能。小胶质细胞吞噬作用在 ICANS 发病机制中的作用以及小胶质细胞表达 CD22 对 CD22CAR T 细胞治疗的后果尚待阐明,但细胞因子介导的小胶质细胞活化在脑型疟疾患儿中已有报道:这些儿童存在弥漫性脑病伴 BBB 破坏和脑水肿,临床特征与 ICANS 无差异。
脑水肿。脑水肿是 CAR T 细胞治疗后一种罕见但潜在致死性神经系统并发症。现有证据表明,脑水肿的病理生理学可能与 ICANS 中更常见的脑病表现不同。在一项评价 CD19CAR T 细胞治疗 B 细胞急性淋巴细胞白血病成人患者的临床试验中,5例患者发生致死性脑水肿而导致试验终止。评估患者特征、预处理治疗和产品属性的根本原因分析中显示,发生脑水肿的患者年龄<30岁,CAR T细胞产品中CD8+ T细胞的百分比较高,CAR T 细胞输注前血清 IL-15 水平较高、血小板水平较低,CAR T细胞群快速扩增,在第1周内达到峰值,与血清 IL-2 和 TNF 水平急剧升高有关。重要的是,对2例患者的尸检显示 BBB 完全破坏,但 CNS 中无活化 T 细胞。尽管这些结果并不明确,但仍表明血脑屏障的破坏和随后的脑水肿可能是由于炎性细胞因子的激增,而非 CAR T 细胞浸润到CNS。而且分析表明,脑水肿可能是由包括患者特征和产品属性的多因素共同作用所致。
CRS 和 ICANS 的临床管理
低级别 CRS 可通过退热药支持性治疗进行管理,但要确保无其他因素导致发热(如感染)。中/重度 CRS 可接受 IL-6R 阻断抗体托珠单抗治疗,伴/不伴糖皮质激素免疫抑制,以及强化支持治疗,包括液体复苏和血管加压药治疗低血压,并根据缺氧需要进行辅助供氧。低级别 ICANS 通常也通过诊断检查和支持治疗进行管理,而重度 ICANS在大多数中心通常是接受糖皮质激素治疗。托珠单抗可显著降低重度 CRS 的发生率,可能是由于发生 CRS 早期 IL-6 即可达峰,并且 IL-6 是下游炎症级联反应的关键介质。
虽然托珠单抗对于 CRS 非常有效,但在大多数 ICANS 中无效,这可能是由于 CRS 和 ICANS 之间的病理生理学差异和/或托珠单抗穿过 BBB 的渗透性较差。事实上,预防性使用托珠单抗可降低重度 CRS 的发生率,但会增加重度 ICANS 的发生率,可能是由于托珠单抗给药后血清 IL-6 水平升高,IL-6 水平升高是由于通过受体阻断阻止其摄取进入外周组织所致。但这些观察结果需要谨慎解释,因为研究并非随机且样本量较小。尽管皮质类固醇可通过BBB,常用于 ICANS,但仍缺乏其对 ICANS 严重程度或持续时间临床获益的确切证据。大多数研究表明,使用托珠单抗似乎并不影响 CAR T 细胞的疗效;而皮质类固醇对 CAR T 细胞疗效影响的数据相互矛盾,部分研究表明无影响,但其他研究显示不良临床结局伴早期进展和死亡风险增加。这一矛盾进一步强调了需要更好地理解 CRS 和 ICANS 的病理生理机制,并制定管理新策略。
其他临床因素的影响。CAR T 细胞治疗后 CRS 和 ICANS 的发作、严重程度和持续时间可能受到宿主、肿瘤和/或治疗相关因素的影响。基线炎症状态较高的患者(定义为 C 反应蛋白、铁蛋白、D-二聚体和促炎性细胞因子水平)发生 CRS 和 ICANS 的风险会增加,这些患者在 CAR T 细胞输注后可能有发生严重炎症反应的倾向。此外严重 CRS 和/或 ICANS 病例也与多种恶性肿瘤(包括白血病、淋巴瘤和多发性骨髓瘤)肿瘤负荷更高相关,这可能与 CAR T 细胞群扩增更大和同步活化相关。CAR T 细胞输注前预处理治疗的强度似乎也会影响毒性的严重程度,更强的治疗方案可能通过诱导更大的清淋而增加重度 CRS 和/或 ICANS 的风险,清淋可消除细胞因子汇(cytokine sinks)并使稳态细胞因子(如 IL-2 和IL-15)水平更高,可用于 CAR T 细胞增殖。有趣的是,高龄并不增加重度 CRS 或 ICANS 风险。
CAR 设计的影响。CAR分子本身的设计可显著影响 CAR T 细胞的增殖和细胞因子谱,从而影响 CRS 和/或 ICANS 的发生率和严重程度。如上所述,携带CD28 共刺激信号域的 CAR T 细胞产物在输注后似乎增殖更快,其数量似乎比用 4-1BB 共刺激信号域改造的产物更早达到峰值。在临床研究中,这与CRS 和 ICANS更早发作以及严重CRS 和 ICAN发生率更高相关。然而到目前为止,尚未对 CD28 与 4-1BB 共刺激结构域的 CAR T 细胞产物进行直接比较。此外患者特征差异(如肿瘤负荷)以及研究间毒性监测和分级的差异,也可能解释 CD28 和 4-1BB 域在药代动力学和不良事件方面的一些差异。
文献证据还表明,改变 CAR 分子的非信号转导结构域,包括铰链区和跨膜区或抗原结合结构域 (scFv),也可影响 CAR T 细胞治疗相关毒性。例如在临床前模型和 I 期临床试验中,改变CD8α铰链和跨膜结构域的长度可减少携带 4-1BB 和CD3ζ信号结构域的 CD19CAR T 细胞的细胞因子生成,并减少其增殖,但仍保留其细胞溶解活性;重要的是在 B 细胞淋巴瘤患者中该产品可获得较高的完全缓解率,仅出现低级别CRS且无ICANS。与Tisagenlecleucel(一种使用高亲和力 scFv 的 CD19CAR T 细胞疗法) 的研究相比,携带低亲和力 scFv 的CD19CAR T细胞治疗急性淋巴细胞白血病儿童患者时抗肿瘤疗效较高,但不会诱导重度CRS。
接受含 CD28 和CD3ζ信号转导结构域(与获批的 CD19CAR T 细胞axicabtagene ciloleucel 相同,但scFv、铰链和跨膜结构域不同)的 CD19CAR T 细胞治疗的 B 细胞淋巴瘤患者的抗淋巴瘤活性相似,但 ICANS 的发生率和严重程度显著较低。在一项 CD19CAR NK细胞的小型研究中,B 细胞恶性肿瘤患者可实现高完全缓解率且无 CRS 或 ICANS,这表明不同的免疫效应细胞可能具有不同的毒性特征。最后,编码CD3ζ中单个免疫受体酪氨酸活化基序(而非其中3个基序)的 CAR 已在临床前研究中显示可增强疗效和记忆 T 细胞分化,而不增加炎症活性。
总之,上述报告表明,降低肿瘤负荷和基线炎症状态、调整预处理方案和优化 CAR 分子和/或 CAR T 细胞产品的设计,可降低 CRS 和 ICANS 的发生率和/或严重程度。
管理CRS和ICANS的新策略
细胞因子分泌过多和失调是 CRS 的病理核心。非特异性免疫抑制剂(如皮质类固醇)在许多情况下可缓解患者症状,此外,预计其他广谱细胞因子抑制剂(如阻断 JAK1 和 JAK2 的芦可替尼或阻断JAK1(细胞因子受体信号转导所需的激酶)的itacitinib)会减弱促炎性细胞因子(如IFNγ和IL-6)的作用。事实上在 CAR T 细胞相关毒性的临床前模型中,芦可替尼和 itacitinib 均可降低毒性和细胞因子分泌。同样,伊布替尼是一种 BTK 抑制剂,也可抑制 IL-2 酪氨酸激酶(ITK;一种参与近端 T 细胞受体信号转导的激酶),导致小鼠模型体内细胞因子分泌减少(包括 IL-6 的分泌),但也会导致 CAR T 细胞来源的细胞因子(如IFNγ)水平降低,可能表明 CAR T 细胞活化减弱。伊布替尼的研究仅在短期内(CAR T细胞转移后第4天)监测抗肿瘤疗效,因此未观察到差异。有趣的是,在与套细胞淋巴瘤细胞系共培养中,伊布替尼可减少肿瘤细胞的细胞因子分泌,证明了这种治疗方法具有广泛作用。小分子激酶抑制剂常可与多个靶点结合,从而提出了一个可能性,即芦可替尼和伊布替尼直接影响 CAR T 细胞活化水平从而影响临床结局。
另一项研究利用激酶抑制剂的不完全特异性,为基于CD3ζ链的 CAR T 细胞创建了一个 “开/关”开关。达沙替尼是一种 BCR-ABL 靶向激酶抑制剂,获批用于治疗各种血液恶性肿瘤,能够以快速和可逆的强效抑制 CAR T 细胞介导的细胞毒性和细胞因子生成;而且在临床前模型中短期给予达沙替尼也可降低 CRS 相关死亡率,且在去除药物和恢复 CAR T 细胞活性后不会损害体内抗肿瘤疗效(图3)。最近的另一项临床前研究表明,通过将磷酸酶 SHP1 改造后募集到免疫突触中来减弱 LCK 信号传导,可减少 CAR T 细胞产生效应细胞因子以及 CRS 的严重程度,从而进一步支持活化 CAR T 细胞在启动 CRS 中的重要作用。此外,BBz CAR(携带 4-1BB 和CD3ζ共刺激结构域)的铰链、跨膜和膜近端胞内结构域结构经过修饰,呈钝化活化和效应细胞因子生成减少的表型,与历史数据相比,在SCID–beige异种移植临床前模型和临床中的毒性均降低。
总的来说,这些研究表明,广谱细胞因子抑制(通过不同机制)可减少 CRS 病理学,基于我们对 CRS 病理生理机制的了解,这一结果是可以预期的。但长期抑制细胞因子可能对 CAR T 细胞的抗肿瘤疗效有害,因此旨在选择性破坏特定细胞因子信号转导通路的靶向干预措施可能更优。在一个CD19CAR T 细胞治疗后重度 ICANS 或噬血细胞性淋巴组织细胞增生症患者的小病例系列(n=8)中,阿那白滞素阻断 IL-1R 似乎对一半患者有获益。明智使用靶向其他细胞因子(如IFNγ,emapalumab)的新型药物,则可能有助于阐明其在重度 CRS 管理中的作用,而未来的临床研究应能够证明此类方法在预防和/或治疗时的获益。
结论和未来方向
目前CAR T细胞疗法的广泛采用,部分受限于需要在 CRS 和 ICANS 等常见毒性有管理经验的中心,以及由此产生的经济和健康负担。而更深入地了解 CRS 和 ICANS 的分子学和细胞病理生理学将促进开发有效的靶向治疗,以降低毒性而不影响抗肿瘤活性。此外新设计的 CAR 结构也可以尽量减少引起 CRS 和 ICANS 的风险,同时优化了肿瘤抗原的识别和有效的 T 细胞信号传导。
目前的另一个局限在于需要更多地了解 CAR T 细胞的生物学和作用机制,其中包括 CAR 的生物物理特性及其共刺激结构域对基因表达谱的影响,从而可能改变 T 细胞亚群、功能、记忆电位和耗竭。为了提高 CAR T 细胞疗法的临床疗效,迫切需要生成具有最佳体内健康度、持久性和有效性的 T 细胞。
除了肿瘤学,在自身免疫和实体器官移植领域,人们对利用基于 CAR 的技术生成抗原特异性调节性 T 细胞也有极大兴趣,它们有可能实现靶向递送免疫抑制。这些调节性 CAR T 细胞的体内生物学和功能将与目前观察到的效应 CAR T 细胞的特性不同,也可能发生一些新的毒性。工程 T 细胞疗法能否在肿瘤学和非恶性适应症中安全和广泛使用,仍取决于能否实现对这些并发症有效和可靠的预防。
参考文献
Emma C Morris, Sattva S Neelapu, Theodoros Giavridis, Michel Sadelain.Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy.Nat Rev Immunol . 2022 Feb;22(2):85-96. doi: 10.1038/s41577-021-00547-6.

