NEJM:慢病毒基因疗法拯救10名“泡泡男孩”,帮助重建免疫系统
时间:2022-12-24 06:00:36 热度:37.1℃ 作者:网络
1971年9月21日,一个名叫大卫·菲利浦·威特(David Phillip Vetter)男孩出生,然而,不幸的是,他患有一种极其罕见的基因缺陷疾病——重症联合免疫缺陷病(简称SCID),从出生那一刻起,他就生活在一个无菌透明的塑料隔离罩中,因此大卫也被称为“泡泡男孩”(Bubble Boy)。
1984年2月22日,与病魔和孤独斗争了12年半的大卫离开了人世。2001年,一部名为《泡泡男孩》的电影上映,让这种罕见病为更多人所知。
重症联合免疫缺陷病(SCID)是一类非常罕见且非常严重的遗传疾病,发病率仅10万分之一,患有SCID的人,体内没有任何免疫系统,因此缺乏任何抵御细菌、病毒的能力。对患者来说,外界充满着致命的威胁,甚至连母亲一个充满疼爱的吻或者拥抱,都可能会给他们带来可怕的后果,往往出生后不久就会因感染而夭折。
2022年12月22日,美国加州大学旧金山分校的研究人员在《新英格兰医学杂志》(NEJM)上发表了题为:Lentiviral Gene Therapy for Artemis-Deficient SCID 的临床研究论文。
这项临床试验通过慢病毒基因疗法成功治疗了10名患有Artemis缺陷型重症联合免疫缺陷(ART-SCID)的儿童。

此前,已有研究团队使用慢病毒基因疗法成功治疗了其他类型的重症联合免疫缺陷病(SCID)患儿。例如Orchard Therapeutics公司使用慢病毒基因疗法治疗了50名腺苷脱氨酶缺陷型重症联合免疫缺陷病(ADA-SCID)患儿。Mustang Bio公司使用慢病毒基因疗法治疗了8名X染色体连锁重症联合免疫缺陷病(SCID-X1)患儿。这两项研究分别于2019年和2021年发表于《新英格兰医学杂志》(NEJM)
然而,Artemis缺陷型重症联合免疫缺陷(ART-SCID)是SCID的一种罕见形式,占全部SCID患者的2%-3%,由编码Artemis蛋白的DCLRE1C基因突变所致,这类SCID患者通常对标准疗法骨髓移植的反应更差,易发生排斥、移植物抗宿主病,从而导致慢性感染和死亡。
参与这项临床试验的儿童目前年龄在18个月到4.5岁之间,其中9人出生在美国,在新生儿筛查后被诊断为SCID,1人出生在加拿大,在5个月大时被诊断出患有SCID。中位随访时间为31.2个月。在该研究发表时,10名患儿全部存活,6名患儿已被随访至少24个月。
这项临床试验的领导者 Morton Cowan 博士表示,自己此前已经使用标准疗法骨髓移植治疗了30多名ART-SCID患儿,但这一次的慢病毒基因治疗的效果要比之前的骨髓移植好多了,从未在之前的治疗中见到这么好的效果,这简直是个奇迹。
例如,接受治疗的一位名为 H.T. Begay 的小男孩,他出生于2018年,出生后不久就发现了患有ART-SICD,他也是这项临床试验的第一个参与者。这项临床试验治愈了他,让他过上了正常的生活,不再需要药物维持,能够在家里的农场中自由快乐地成长。
在这项临床试验中,研究团队从患儿的骨髓中提取干细胞,然后在体外使用慢病毒载体将正确版本的DCLRE1C基因导入,体外扩增后再将这些基因工程改造后的细胞回输到患儿体内。整个过程与CAR-T细胞疗法类似。
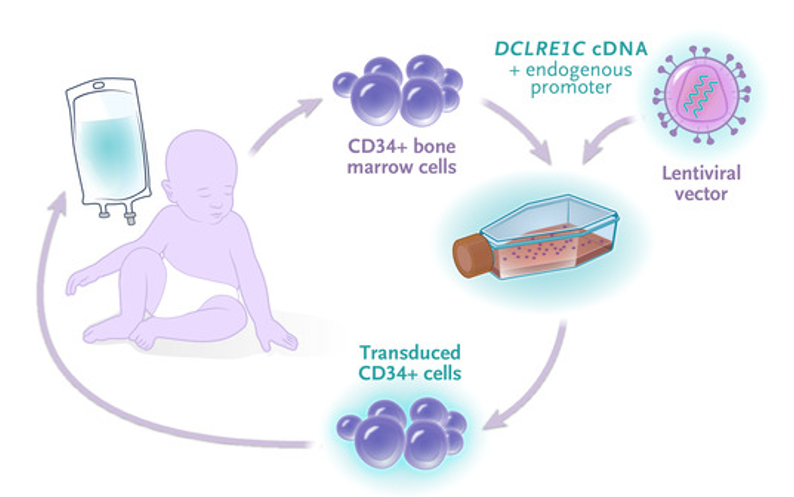
所有10名患者都被安全地输入了他们自身来源的经过基因工程改造的的干细胞,这些干细胞在42天内产生了正确的外周血细胞。在12周时,所有10人都生长出了自己的T细胞和B细胞,9人(有1人接受了第二次治疗)中有4人在12个月时实现了完全的T细胞免疫重建,并在24个月时实现了完全的B细胞免疫重建,这使他们能够停止免疫球蛋白替代治疗,并能够接受标准的儿童疫苗接种。还有3人随访时间少于24个月,他们与之接受骨髓移植的患儿相比,有更好的B细胞发育。
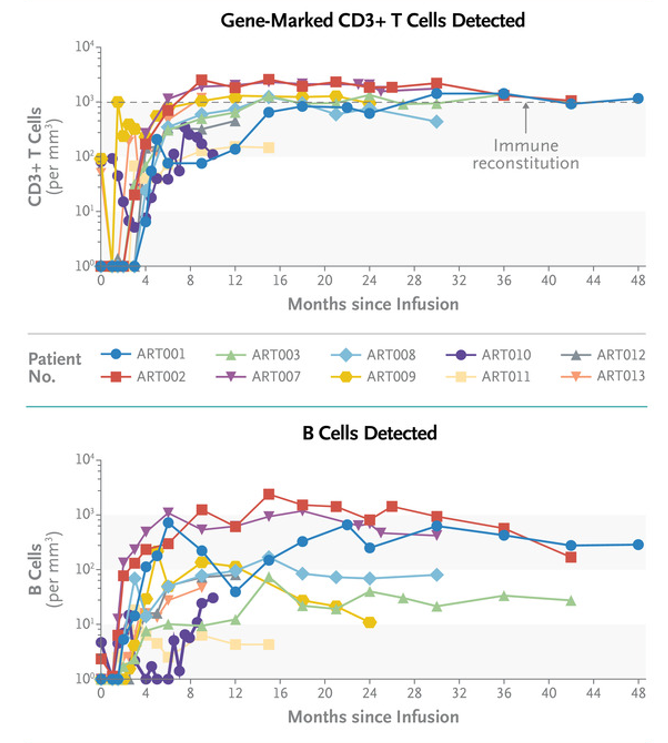
1名患儿因为在基因治疗前持续感染巨细胞病毒,进一接受了第二次输注治疗,现在他已无感染,且具有了良好的T细胞B细胞,所有的结果都比之前接受骨髓移植的ART-SCID患者更好。
这项临床试验的领导者 Morton Cowan 博士表示,这项基因疗法帮助患儿建立了更好的B细胞免疫,这有助于他们避免慢性肺部疾病问题,这类疾病通常发生在接受标准疗法骨髓移植的ART-SCID患者的童年后期。
原始出处:
Morton J. Cowan, et al. Lentiviral Gene Therapy for Artemis-Deficient SCID. N Engl J Med 2022; 387:2344-2355.

