EMBO J:Jacob可作为抗阿尔茨海默病突触功能障碍的靶点
时间:2023-04-15 09:14:37 热度:37.1℃ 作者:网络
可溶性β-淀粉样肽(Aβ)引起的突触功能障碍是早期阿尔茨海默病(AD)的标志,与认知能力下降密切相关。通过未知的机制,Aβ抑制camp反应元件结合蛋白(CREB)的转录活性,CREB是细胞存活和可塑性相关基因表达的主要调节因子。近期《The Embo Journal》期刊发表了题为“Jacob-induced transcriptional inactivation of CREB promotes Aβ-induced synapse loss in Alzheimer’s disease”的文章,报道了Aβ在AD患者的大脑和小鼠海马神经元中诱导了Jacob的核胞质转运,Jacob是一种连接nmda受体衍生信号体与CREB的蛋白。Aβ调节Jacob的运输诱导CREB的转录失活,导致AD小鼠模型的突触损伤和丢失。小化合物硝苯胂酸选择性地阻碍Jacob/ LIM-only 4 (LMO4)/蛋白磷酸酶1 (PP1)信号体的组装,从而恢复CREB的转录活性。硝苯胂酸可预防AD小鼠模型突触可塑性损伤和认知能力下降。总的来说这些数据表明靶向Jacob蛋白诱导的CREB失活可作为对抗AD早期突触功能障碍的治疗途径。

我们首先检测了AD患者死后组织中pJacob和pan Jacob的水平(患者信息见表EV1),为该蛋白在人类AD病理中的潜在参与提供证据。与对照组相比,AD患者颞皮质核富集部分的免疫印迹并没有显示pan Jacob水平的显著降低(图EV1A-C)。然而,pJacob的水平显著降低了约40%(图1A、B和EV1A),这表明突触NMDAR激活后Jacob的核输入减少,可能是以突触外NMDAR激活为代价的。
意料之中的是,我们通过NeuN-免疫印迹法在AD患者中观察到明显的神经元损失(图EV1D和E)。由于Jacob与CREB不同,仅在神经元中检测到,我们可以将核Jacob的磷酸化归一化到总蛋白水平,并校正这些NeuN含量的值,以调整神经元细胞损失(图1C)。通过这种方法,我们观察到pJacob/Jacob比值的明显降低(图1C),这与CREB失活的程度相关,这是我们在对neun阳性核(图1D-F和EV1F)进行FACS分选后确定的,因为CREB在胶质细胞和神经元中都有表达。综上所述,这些数据表明人类AD大脑中的CREB失活。此外,低水平的pJacob表明CREB失活和突变之间的功能联系是由Jacob介导的。
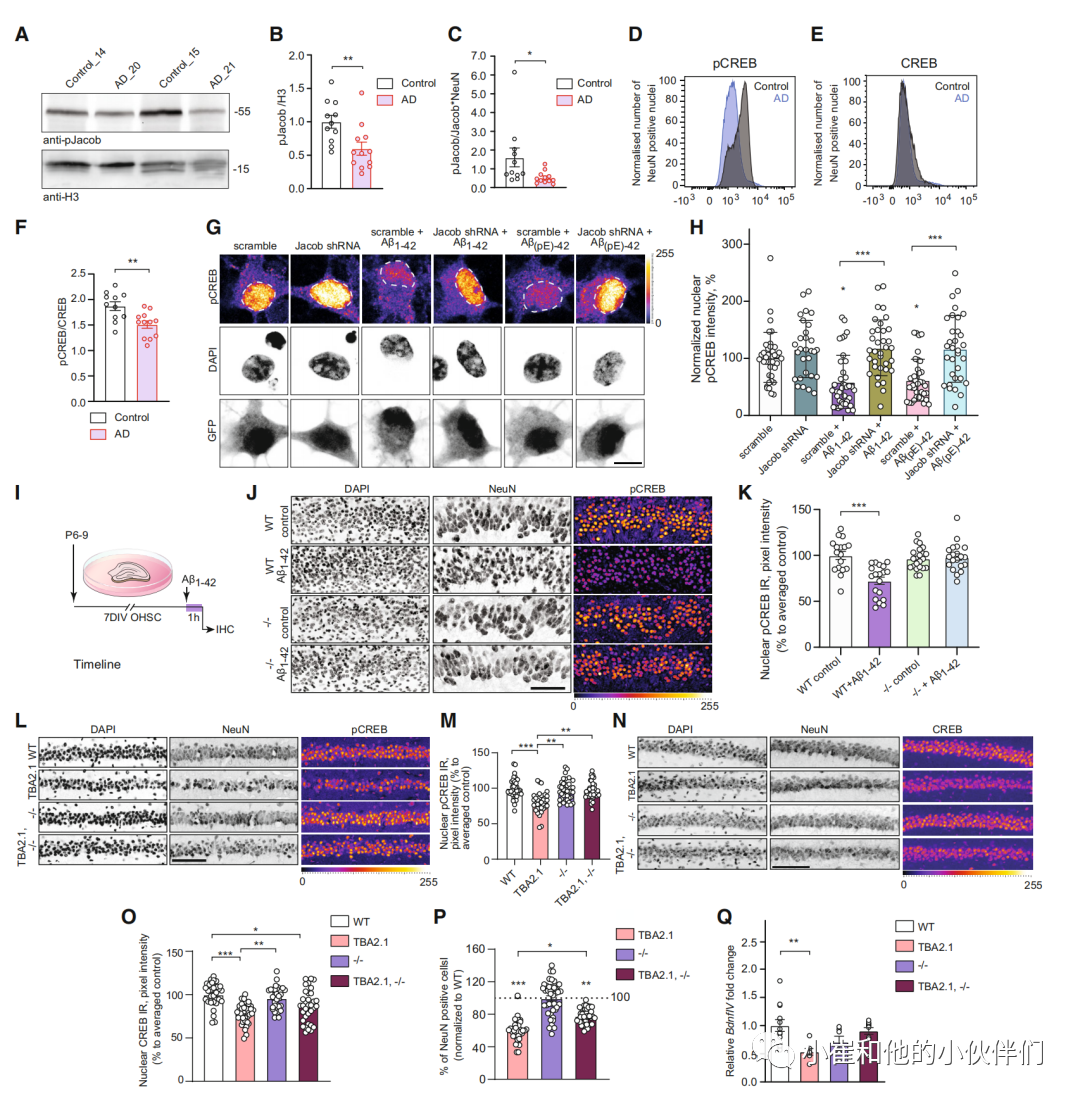
图1.Jacob在人类阿尔茨海默病(AD)和AD小鼠模型中与camp响应性元素结合蛋白(CREB)失活有关
Aβ低聚物可以以各种翻译后修饰的形式存在,其中n端截断的焦谷氨酰化Aβ3(pE)-42物种在AD患者的大脑中表现突出。先前的研究表明,Jacob可能在Aβ诱导的CREB失活中发挥作用,这种失活是由包含NMDAR的突触外GluN2B激活引起的,机制尚不清楚 。在海马神经元中shRNA敲除Jacob确实阻止了500nmAβ1-42或Aβ3(pE)-42低聚物培养物处理诱导的CREB失活(图1G和H)。在Jacob/Nsmf敲除小鼠(-/-)或野生型同巢小鼠的有机型海马切片中也获得了类似的结果(图1I-K)。两种基因型的pCREB基础免疫荧光水平无差异;然而,与野生型小鼠不同,敲除小鼠的神经元没有出现Aβ1 -42诱导的CREB失活(图1J和K)。总CREB水平保持不变(图EV1G和H)。
我们接下来推断,在Jacob敲除小鼠中缺乏Aβ诱导的CREB失活可能对AD具有神经保护作用。海马体的CA1区是AD最早受影响的区域之一,具有明显的神经元损失和突触接触数量减少。TBA2.1小鼠表达Ab3(pE)-42,并很早就表现出严重的CA1神经元丢失、淀粉样变、LTP损伤和神经炎症。我们选择TBA2.1小鼠是因为它们可能在所有转基因AD小鼠模型中表现出最具侵略性和最突出的淀粉样蛋白病理。
Western blot分析TBA2.1小鼠大脑蛋白提取物显示,Jacob蛋白水平保持不变,pJacob水平降低导致pJacob/Jacob比降低,与人脑所得的结果一样(图EV1I-L)。根据来自其他AD转基因小鼠系的报道,我们发现TBA2.1小鼠表现出显著降低核pCREB水平(图1L和M)。为了直接研究神经元中Jacob表达的缺失是否能在TBA2.1小鼠中提供神经保护,我们接下来杂交小鼠,以获得纯合子TBA2.1和Jacob/Nsmf/)老鼠。有趣的是,双转基因动物(TBA2.1 × Jacob/Nsmf -/-)没有显示出CREB失活,这证明pCREB水平没有降低(图1L-O)。虽然我们在所有三种基因型(TBA2.1, Jacob/Nsmf -/-,双TBA2.1 × Jacob/Nsmf -/-)中观察到核CREB水平略有下降(图1N和O),但Jacob/Nsmf -/-和双转基因动物中CREB失活的缺失表明该蛋白在阿巴淀粉样变早期转录失活的关键作用。因此,双转基因小鼠背侧CA1区域的细胞损失不太明显(平均23%)(图1P)。
当我们使用SPECT对四种基因型(TBA2.1, Jacob/Nsmf (-/-), TBA2.1 × Jacob/Nsmf-/-和野生型(WT))无约束行为小鼠的脑血流(CBF)成像时,发现Jacob基因敲除介导的拯救在全脑网络激活模式水平上也可见。与野生型动物相比,TBA2.1小鼠背侧CA1(箭头)中发现的CBF减少,在双转基因小鼠中得到部分挽救(图EV1M)。侧隔和对角带(连接海马的区域)也有明显的缓解(图EV1N)。此外,BDNF基因启动子IV (BdnfIV)转录的BDNF mRNA水平在TBA2.1小鼠中下降,但在Jacob/Nsmf (-/-)和双转基因动物中没有下降(图1Q), BdnfIV是一种突触可塑性相关的神经营养因子 ,其表达受CREB以活性依赖的方式调控。
Jacob基因缺失不影响星形胶质细胞的数量(图EV1O和P)和活化的小胶质细胞(图EV1O和Q)。此外,aβ阳性沉积的数量(图EV1R和S)所证明的淀粉样蛋白负荷也不受影响,这表明神经炎症或淀粉样蛋白沉积的间接作用并不能解释Jacob基因缺失所赋予的神经保护作用。
这些数据共同表明,jacob诱导的CREB失活(我们称之为JaCS)有助于AD中CREB的转录失活,因此我们接下来的目标是破译潜在的分子机制。我们首先测试了两种蛋白质之间可能的直接相互作用。细菌表达蛋白的下拉实验显示,Jacob的n端117-172氨基酸(aa)和c端(262-532 aa)区域与CREB的bZIP结构域直接相关(图2A和EV2A-C)。
因此,超分辨率刺激发射耗竭(STED)成像显示,在培养的海马神经元中,核Jacob靠近CREB(图EV2D和E)。在异源表达Jacob(图EV2F)后,我们可以从HEK293T细胞中共免疫析出内源性CREB(图EV2F),为了支持这些数据,我们发现当我们共表达全长或n端半段Jacob和CREB的c端片段时,体内荧光共振能量转移(FRET)效率显著(图EV2D和E)2中)。值得注意的是,Jacob的n端片段比c端片段产生了明显更强的FRET信号(图2B - F)。最后,近距离结扎实验为Jacob与CREB在海马原代培养神经元核中的相互作用提供了证据(图2G)。
在以Jacob终止子诱导的酵母双杂交(YTH)筛选中,明确了LMO4为结合伙伴(附录图S1A)。LMO4是CREB的转录共激活因子,因此我们想知道Jacob-LMO4相互作用是否在JaCS中起作用。Jacob的a117 -228区域与LMO4的LIM1结构域直接相互作用(图2H,附录图S1A和B)。
此外,这两个蛋白在神经元核中相互靠近通过STED成像(附录S1C和D)和LIM1结构域与异源表达后的细胞质Jacob簇共定位(附录S1E)。通过与标志特异性抗体的异源免疫共沉淀进一步证实了这两种蛋白的相互作用(附录图S1F),体内FRET分析证实了直接相互作用(图2I-K),海马神经元的近距离连接表明存在体内相互作用(图2L)。随后的Jacob片段与LIM1结构域的异体共免疫沉淀表明LMO4和CREB的结合区域不重叠(图2F和M,附录图S1G)。有趣的是,像其他LIM1结构域结合蛋白一样,Jacob含有富含亮氨酸和缬氨酸的延伸。该区域内的点突变(L175A-V176A)导致结合更弱(图2M-O)。在之前的工作中,我们发现非磷酸化Jacob的核过表达导致CREB去磷酸化。然而,Jacob的LMO4结合突变体的核积累并没有诱导CREB失活(图2 - t),这表明与LMO4的关联系是JaCS的工具。
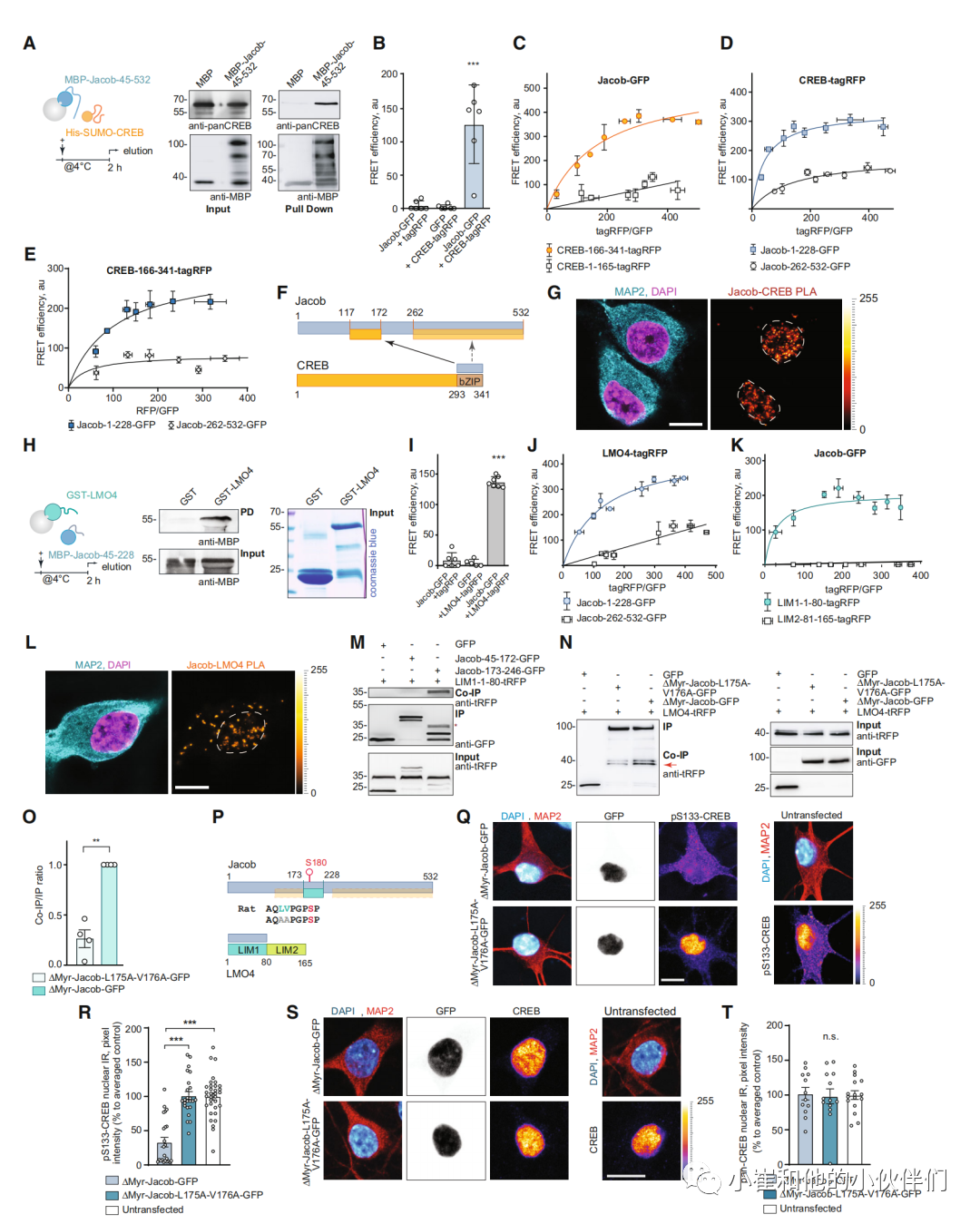
图2.Jacob直接与camp反应元件结合蛋白(CREB)和LMO4结合
我们接下来研究为什么与LMO4的联系对JaCS至关重要。体内FRET试验和异源共免疫沉淀显示,LIM1结构域是Jacob的结合界面(图2J, K, M和P,附录图S1A, E和F)也与CREB的n端片段相互作用(图3A-C)。LMO4直接与该区域结合,而不与CREB的孤立bZIP结构域结合(图3D-F,附录图S1B和G),从而与Jacob结合。与LMO4的直接相互作用促进了S133的磷酸化,从而促进了CREB的转录活性,这可以通过异源表达后CREB驱动的荧光素酶活性的增加证明LMO4的作用(图3G)。神经元中shRNA敲低LMO4(附录图S1H-J)会导致pCREB减少(图3H和I),但不会导致全部CREB免疫荧光减少(图3J和K)。
LMO4与CREB的直接结合,Jacob引出了这三种蛋白质是否可以在三聚体复合体中组装,或者它们是否可以竞争相同的结合界面的问题。在体内的序列共振能量转移(SRET)确实揭示了一个三重复合物的存在(图3L和M)。Jacob在n端有一个与CREB和LMO4相互作用的结合界面(图2),而LMO4只能通过其第一个LIM1结构域与Jacob或CREB相关联(图2和3)。随后的竞争下拉实验证实了Jacob和CREB n端与LMO4之间的竞争结合。当GST-LMO4偶联到珠粒上,在CREB存在的情况下增加Jacob的n端片段(图3N-P)。总之,LMO4的LIM1结构域介导了与CREB或Jacob的结合,而Jacob能够取代CREB复合体中的LMO4,这一机制应该有助于CREB失活(图3Q)。
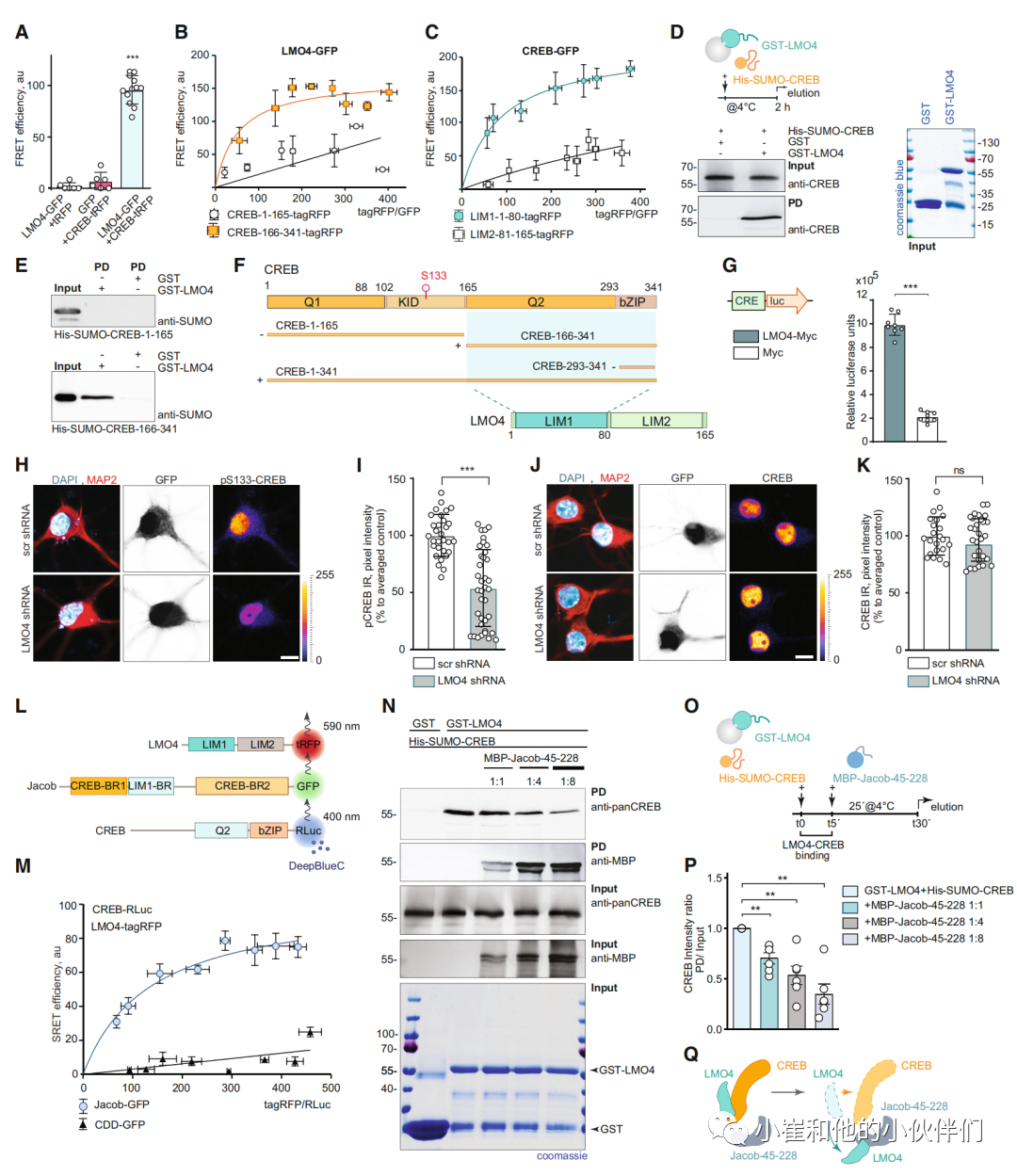
图3.Jacob在camp反应元件结合蛋白(CREB)中取代LMO4
我们接下来考虑lmo4与Jacob的结合是否也可能积极参与CREB失活。蛋白磷酸酶1 ( PP1)是一种能够在CREB中去磷酸化S133的磷酸酶。Jacob含有多个PP1结合基序,这两个蛋白在异源表达后共定位(附录S2A), Jacob与内源性PP1c共免疫沉淀(附录S2B)。下拉实验显示了直接的相互作用(图4A,附录图S2C),异源免疫共沉淀实验指出了两个结合界面(图4B)。因此我们接下来探讨了与PP1γ的关联是否涉JaCS。为此,我们在海马原代神经元细胞核中表达了一个Jacob的磷酸缺乏突变体 ,并用PP1γ抑制剂软脂肪酸 (OA)培养。有趣的是,我们发现OA治疗可预防JaCS(图4C和D)。
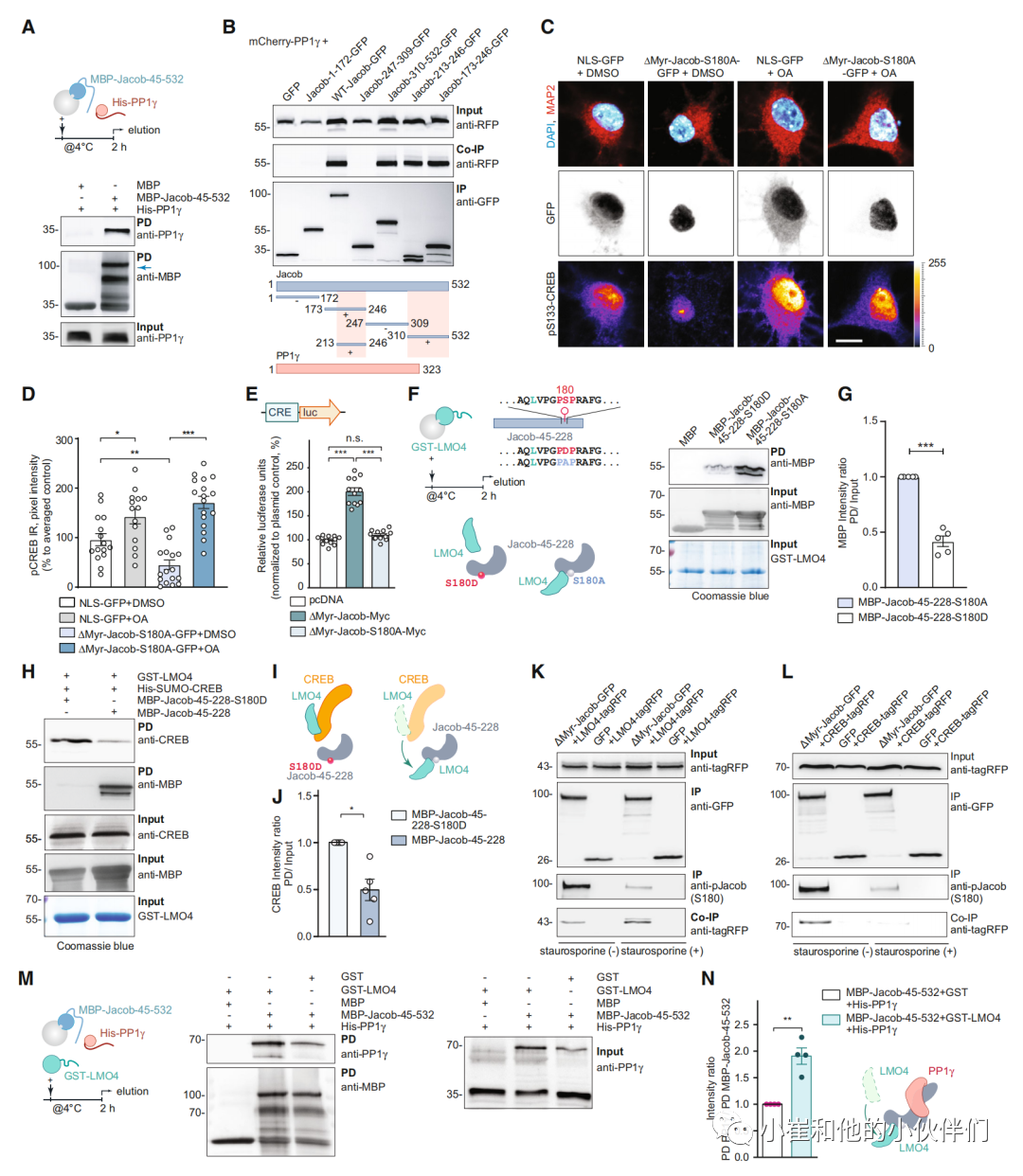
图4.JaCS需要Jacob与PP1γ绑定
Jacob在S180位点的磷酸化是由突触NMDAR激活诱导的,而非磷酸化Jacob的核输入则与突触外NMDAR激活有关 。因此,在cre -荧光素酶活性测定中,野生型Jacob的表达导致活性增加(图4E,附录图S2D)。此外,缺磷突变体Jacob与LMO4的关联明显强于相应的拟磷蛋白(图4F和G,附录图S2E和F)。与未磷酸化的蛋白不同,拟磷Jacob没有将CREB从LMO4上转移到珠粒上(图3N-Q和4H-J)。为了进一步验证LMO4能更有效地与非磷酸化Jacob结合,我们应用了蛋白激酶抑制剂星孢菌素,并进行了异源免疫共沉淀实验(图4K和L)。
我们确实发现非磷酸化Jacob与LMO4有更强的关联(图4K和L),同时S180磷酸化Jacob与CREB有更强的关联(图4K和L)。由于Jacob与PP1γ直接相互作用(图4A),我们进行了下拉试验,观察到在LMO4存在时,两种蛋白之间的关联要强得多(图4M和N,附件图S2E和F)。在突触外NMDAR激活后进入细胞核,会取代CREB复合体中的LMO4,随后与LMO4的结合增强其与PP1γ的结合,最终导致CREB失活(图4I)。
上面概述的分子分析使我们能够对CREB、Jacob和LMO4的LIM1结构域之间的结合界面进行结构建模。为此,我们分析了沉积的肽结合LMO4结构。在LMO4:LIM结构域结合蛋白1 (Ldb1) 中,Ldb1的肽段通过短的b-b主链形成和单疏水侧与LMO4结合在两个LIM结构域中都有突出的链进入深袋(图5A)。LMO4:Ldb1突出了肽与LIM结构域结合的相对较弱的序列偏好(图5A)。
因此,我们通过计算序列突变Jacob和CREB中的每个肽残基到20个aa区段中的任何一个,进行了位置扫描,这让我们为潜在的lmo4结合区域定义了仅四个关键残基的搜索模式(图EV3A-D)。通过这种方法,我们在Jacob中发现了8个潜在的结合区域,在CREB中发现了5个(图EV3B),为此我们模拟了LIM1:肽复合物,并计算了复合物的稳定性。对于Jacob,我们确认了一个包含172-186残基的肽,它是实验定位的LMO4结合区域的一部分。此外,我们在实验确定的CREB的lmo4结合区域内发现了两个重叠的肽(图EV3D)。
接下来,我们寻找可能选择性地阻止Jacob而不是CREB与LMO4的LIM1结合的小分子。在这里,我们使用了ZINCPharmer,这是一种允许定义LIM1疏水结合口袋(图5B)和β链(图5C)内的给体和受体原子的工具 。一些结果含有一个对硝基苯基团(例如,2-[[1-(4-硝基苯)乙基]氨基]ethan-1-ol (ZINC37177221)),与疏水结合袋相吻合。因此,我们接下来搜索了Drugbank数据库,以寻找含有该基团的药物,并将对硝苯胂酸(Nitarsone)确定为候选药物,因为AutoDock Vina证明硝苯胂酸适合LIM1的防水结合袋 。
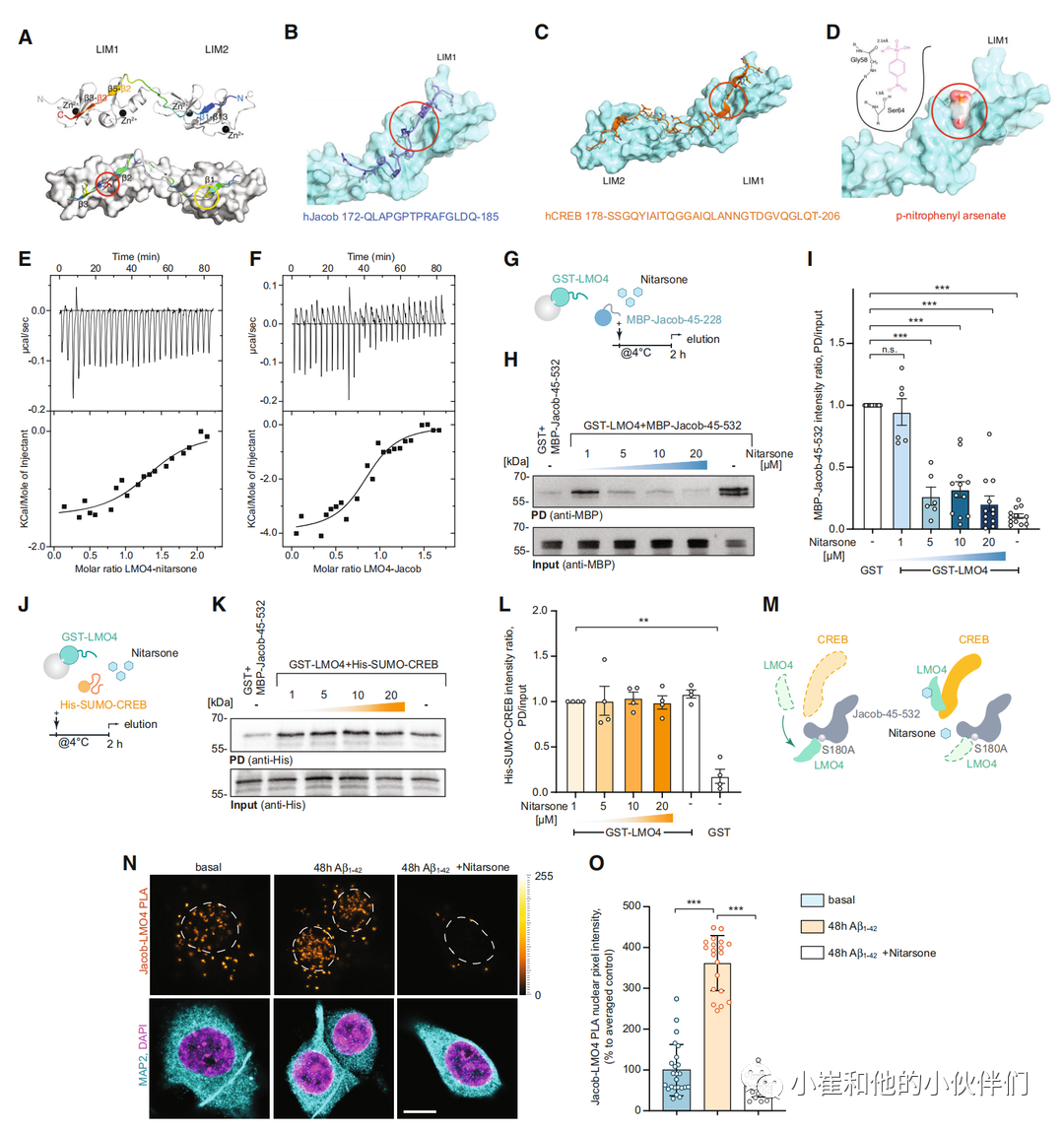
图5.硝苯胂酸破坏Jacob的lim1结构域结合
为了证明硝苯胂酸与LIM1结构域结合的有效性、特异性和亲和力,接下来纯化了在细菌中表达的重组蛋白(图EV3E)。等温滴定量热法(ITC)显示LMO4中有一个单一的结合位点,KD为0.77uM(图5E),这与Jacob与LMO4结合的KD为0.37 uM大致吻合(图5F)。
这些结果验证硝苯胂酸只阻断LMO4与Jacob的结合,而不阻断CREB的结合的预测。在GST下拉实验中,我们结果表明,在5倍摩尔过量的用量下,硝苯胂酸完全消除了Jacob与LMO4的结合(图5G-I和EV3E)。在这些实验中,我们固定化500 nM的GST-LMO4,用1uM Jacob 45-228饱和结合,然后应用5uM 硝苯胂酸。相反,即使浓度为20 uM,即20倍摩尔过量,该物质也没有从LMO4中置换CREB(图5J-M和EV3E)。最后,我们使用近距离结扎法测试了5 uM硝苯胂酸对海马原代培养Jacob-LMO4复合物形成的影响。正如预期的那样,硝苯胂酸大大减少了Aβ诱导 Jacob与LMO4在神经元中的关联(图5N和O)。
接下来,我们发现10 uM硝苯胂酸可防止Aβ诱导的海马初级神经元CREB失活(图EV4A-C)。用药前30分钟或用药后2小时,500 nM Aβ持续2小时起作用。在这两种情况下,我们发现硝苯胂酸给药后pCREB水平都得到了挽救(图EV4A-C),这表明该药物不仅会阻止Aβ治疗对Jacob的反应,而且即使在Aβ诱导的CREB失活后,也会取代与LMO4结合的Jacob。联合应用5 uM 硝苯胂酸,这个剂量已经有效地中断了初级神经元中的Jacob/LMO4相互作用(图5N和O),也挽救了500 nM Aβ低聚物48小时诱导的CREB失活(图6A和B)。
因此,我们接下来评估了在5 uM硝苯胂存在下保存48小时的游离海马神经元中Aβ诱导的突触丧失(图6 c和D)。Aβ诱导可减少30% 突触小泡蛋白(图6 c和D)但硝苯胂酸完全阻止这种突触损失(图6 c和D)。GluA1 AMPA受体突触表面表达的下调也显著减弱((图6 e和F)。在对照实验中,这些神经元表现出正常的GluA1 AMPA受体增加,以响应1 uM TTX应用对神经元活动的沉默(图EV4D)。全细胞膜片钳实验显示,Aβ处理后GluA1表面表达降低,伴随突触后微兴奋性电流(mEPSC)振幅降低,但频率不降低(图6G-K)。联合应用5 uM硝苯胂酸可保持mEPSC振幅,而单独给药对两种测量均无影响(图6G-K)。
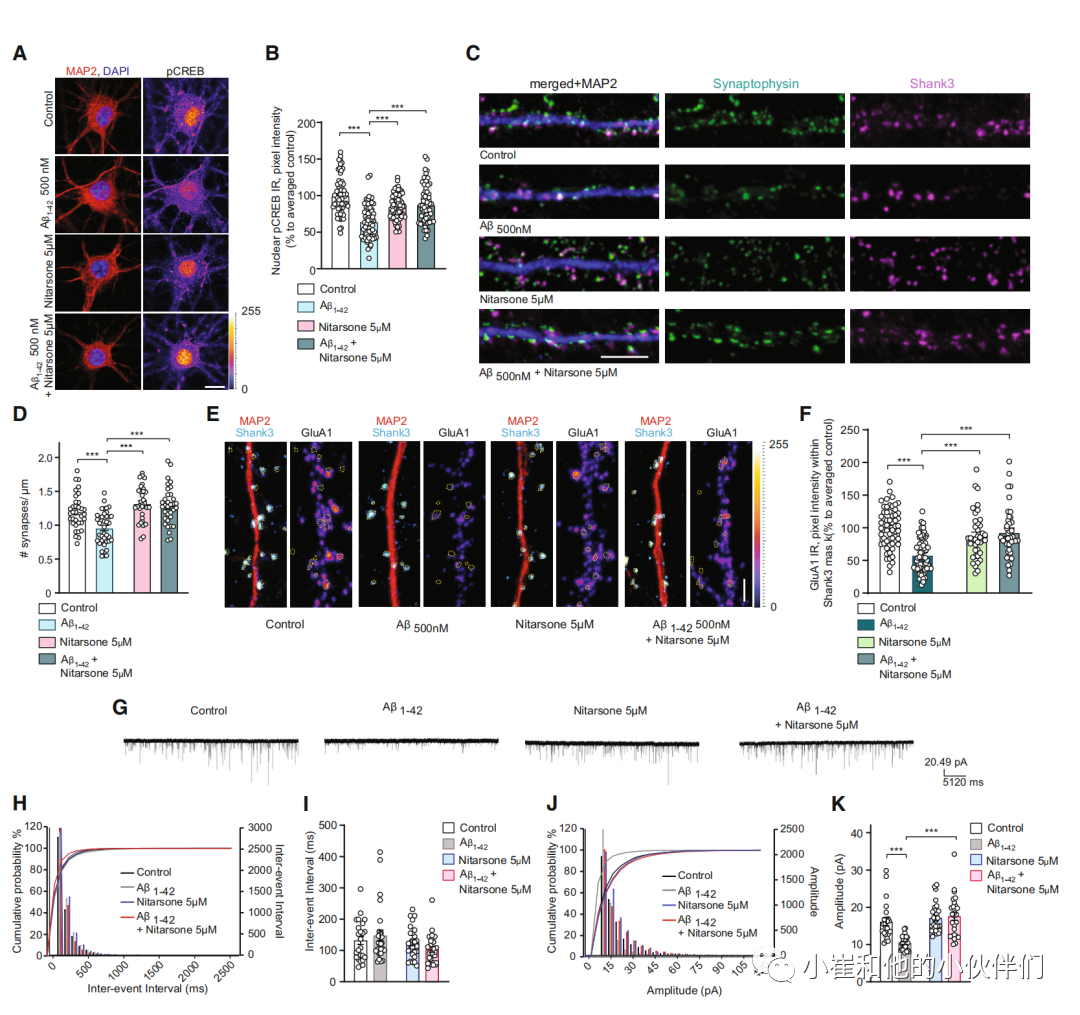
图6. 硝苯胂酸治疗可挽救Aβ1 -42诱导的突触功能障碍
接下来,我们在两个淀粉样蛋白病理的转基因AD小鼠系TBA2.1和5xFAD小鼠体内给予硝苯胂酸。5xFAD小鼠表达人类APP和PSEN1转基因,共有5个ADlinked突变 。与TBA2.1小鼠相比,这些小鼠表现出更慢的淀粉样蛋白病理扩散,在4个月龄时伴有胶质细胞增生的可见斑块,并伴有突触功能障碍和认知障碍。小鼠喂食硝苯胂酸,每日剂量为50 mg/kg,这是基于几项毒理学研究的保守NOEL(未观察到影响水平)和在脑组织中达到有效剂量的基本原理(实验细节见材料和方法和图7A)。
与对照动物相比,该方案对治疗动物的体重没有影响(图EV5A和B),对两种小鼠系的淀粉样蛋白负荷也没有影响(图EV5C-F)。然而,硝苯胂酸在治疗6周后有效地防止了两种转基因AD小鼠海马背侧CA1区域的CREB失活(图7B-E和EV5G-J)。此外,与对照处理的同窝小鼠相比,TBA2.1小鼠的早期神经元细胞损失减少(图7F和EV5K和L),而我们在19周龄时无法检测到5xFAD小鼠CA1中的神经元细胞损失(图EV5M和N)。最重要的是,我们发现在两种淀粉样蛋白病理动物模型中,硝苯胂酸处理的小鼠的陷窝层分子突触损失明显减少(图7G-J)。在许多AD动物模型中,这是第一个受到淀粉样蛋白病理影响的海马区域 。图为,该区域的突触消失现象。树突静脉曲张对TBA2.1小鼠的影响(图7K和L),这是AD病理一个形态学特征 。硝苯胂酸治疗小鼠的树突静脉曲张明显减少(图7K和L)。
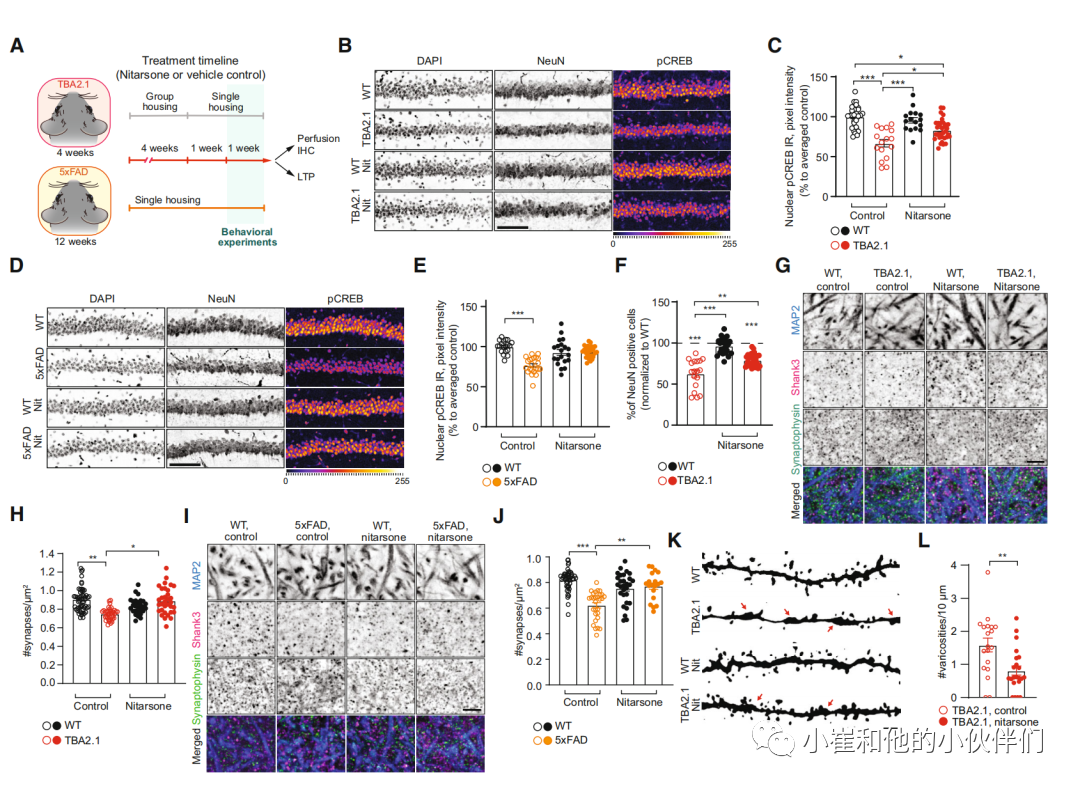
图7. 口服硝苯胂酸可挽救TBA2.1和5xFAD小鼠 CREB失活和突触丢失
与这些发现一致,我们分别在出生后第11周和第19周观察到TBA2.1和5xFAD小鼠的急性海马切片中出现早期突触功能障碍,并以晚期长期增强(LTP)缺陷为证(图8A-D)。在最后30分钟的记录中,在两个AD系的硝苯胂酸喂养的小鼠切片中,fEPSP斜率的降低得到了恢复(图8A-D和EV5O和P),这表明与学习和记忆相关的突触可塑性得到了恢复(图8A-D和EV5O和P)。高CREB水平已被证明可以增加外侧杏仁核和海马体神经元的兴奋性。因此,我们假设硝苯胂酸通过预防TBA2.1小鼠JaCS可以增加海马CA1神经元的兴奋性。利用膜片钳记录,我们发现基因型(转基因与野生型)在阶进电流方案中对神经元兴奋性有轻微影响(图8F),这可能反映了这些小鼠AD病理的早期阶段。早期硝苯胂酸处理增加了输入电阻(图8E和F),降低了流变基(图8E),恢复了输入电阻与动作电位半宽的正相关关系(图8),这是在对照条件下的野生型小鼠中观察到的(图8E-G)。后一个观察结果特别有趣,因为在CA1神经元群中,硝苯胂酸处理具有高输入电阻和高半宽的神经元可以特异性地克服突触传递和突触可塑性(LTP)的诱导缺陷。
因此,我们接下来确定硝苯胂酸治疗是否也能挽救TBA2.1和5xFAD小鼠的认知衰退。为了评估短期记忆,我们使用了y迷宫物体识别任务,该任务最小化上下文线索,训练和测试之间的间隔为3小时。与同窝对照组相比,TBA2.1和5xFAD处理小鼠的物体识别能力受损(图8H-J)。相反,转基因TBA2.1和5xFAD小鼠喂食硝苯胂酸显示出更好的辨别性能相比,处理的动物(图8H-J)。人类AD患者表现出物体识别障碍,这主要依赖于CA1神经元的适当突触功能。相应地,TBA2.1和5xFAD小鼠在新物体定位和新物体识别记忆方面表现出缺陷(图8K-M和EV5Q-T)。硝苯胂酸治疗也在新位置识别和新物体识别任务中挽救了记忆,其认知表现与车辆处理的一窝同伴对照组相当(图8N和O)。我们没有观察到行走距离和数量的重大差异训练过程中与对象的交互(即偏好指数)(图EV5Q-T)。总的来说,这些数据证明,尽管存在明显的淀粉样蛋白病理,但硝苯胂酸恢复突触可塑性和正常兴奋性可以改善海马体依赖的学习和记忆。

图8. 口服硝苯胂酸可拯救TBA2.1和5xFAD小鼠的
阿尔茨海默病(AD)相关表型
尽管有强有力的证据表明Aβ驱动的信号通路是AD的病理机制的基础,但近年来,人们怀疑其他途径是否会导致疾病进展和认知能力下降。抗淀粉样蛋白疗法的临床试验未能在AD患者的认知方面显示出强有力的有益结果,这引起了人们的担忧,即降低淀粉样蛋白负荷足以阻止疾病进展。我们推测,鉴于AD所谓的“细胞期”机制的复杂性,以及由于低聚Aβ物种的释放,用抗体溶解β-淀粉样斑块可能会产生有害的副作用,抗Aβ抗体(如Aducanumab)和靶向JaCS的组合干预可能是减缓AD早期突触衰竭的更有效手段。这一假设可以在未来转基因AD小鼠模型的工作中得到验证。
编译仅做参考,精读请下载全文。
Grochowska KM, Gomes GM, Raman R, Kaushik R, Sosulina L, et al. Jacob-induced transcriptional inactivation of CREB promotes Aβ-induced synapse loss in Alzheimer's disease. EMBO J. 2023 Feb 15;42(4):e112453. doi: 10.15252/embj.2022112453.

