【协和医学杂志】一例PTEN新生杂合突变患儿临床表型与免疫特征分析
时间:2023-09-20 17:11:19 热度:37.1℃ 作者:网络
PTEN基因种系突变引起的一系列疾病即为PTEN错构瘤综合征(PHTS),包括考登综合征(CS)、 Bannayan-Riley-Ruvalcaba综合征(BRRS)、成人莱尔米特-杜克洛病(LDD)和与巨脑相关的孤独症谱系障碍(ASD)[1]。
PHTS患者临床异质性显著,即使同一家族具有相同突变的患者,其临床表现也有较大异质性[2]。既往研究认为,PTEN基因突变多累及大脑(如巨脑、精神发育迟缓、ASD等)、皮肤(如多发性毛根鞘瘤、肢端角化等)、甲状腺(如甲状腺腺瘤、甲状腺结节等)、消化道(如肠道息肉),伴多系统肿瘤风险增加[1]。
Tsujita等[3]报道PTEN杂合突变可导致PI3Kδ过度活化综合征(APDS)样表型,本例患儿为PTEN c.388C>T(p.R130X)新生杂合突变,目前尚无此位点突变患儿的免疫学表型报道,因此本研究旨在探讨该患儿临床表型及免疫特征,以丰富PTEN突变相关临床表型谱,提高临床医生对该疾病的认识。
1 病例资料
患儿,男,1岁4个月,因“发现头围大1年余”于2020年10月首次就诊于重庆医科大学附属儿童医院。患儿出生后即发现左上前臂内侧疣状表皮痣,呈线状排列(图1A),皮肤镜检查提示镜下淡红色背景,黄褐色、淡红色均质样结构,“乳头瘤”样结构,见点球状出血性结构,不除外疣状痣。
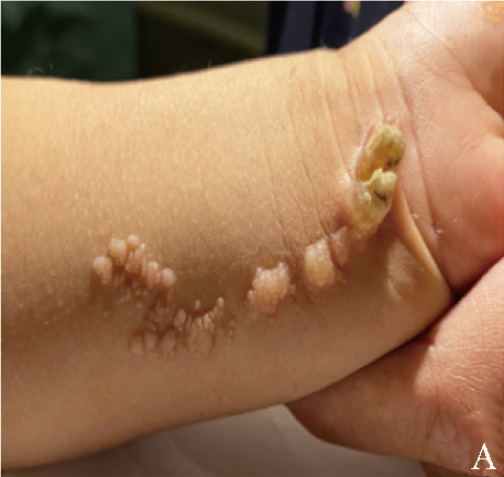
图1A 患儿左上前臂内侧疣状表皮痣
出生后27 d于儿保科体检时发现头围过大(40 cm,>P99),而后多次随访头围均>P99(图1B~1C)。1岁时于外院诊断为精神运动发育迟缓、学语延迟。


图1B 患儿头颅正面照;图1C 患儿头颅侧面照
既往史:新生儿黄疸、新生儿败血症、新生儿结膜炎、双侧睾丸鞘膜积液、毛细支气管炎、佝偻病、鹅口疮、鼻窦炎、脑髓鞘化欠佳、急性支气管炎、急性腹泻、病毒疹。
胸部X线检查示:两肺纹理增重模糊,未见明显斑片状阴影,肺门不大,考虑支气管炎。
腹部超声示:肝脏稍大(肋下2 cm)。
颅脑MRI平扫示:双侧侧脑室体后旁脑白质异常信号、考虑髓鞘化欠佳、枕大池囊肿、鼻窦炎(图1D)。
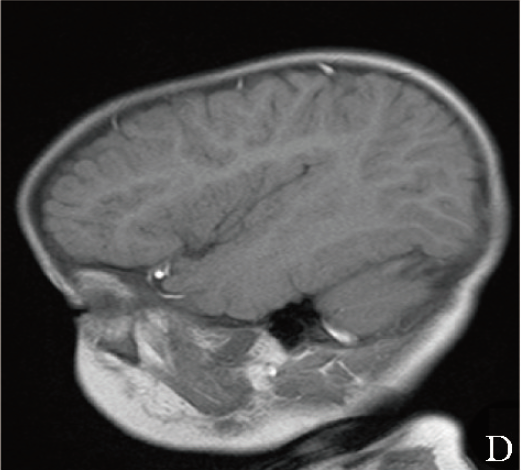
图1D 患儿颅脑MRI矢状位
征得患儿父母知情同意后,取患儿及其父母4 mL肝素抗凝全血送贝瑞基因进行医学全外显子组综合检测,Sanger测序验证患儿及其父母相关基因的突变位点,结果显示:患儿PTEN基因(NM_000314.8)c.388C>T(p.R130X)杂合突变,Sanger测序验证患儿此位点存在杂合突变,患儿父母均未携带此突变基因(图2)。
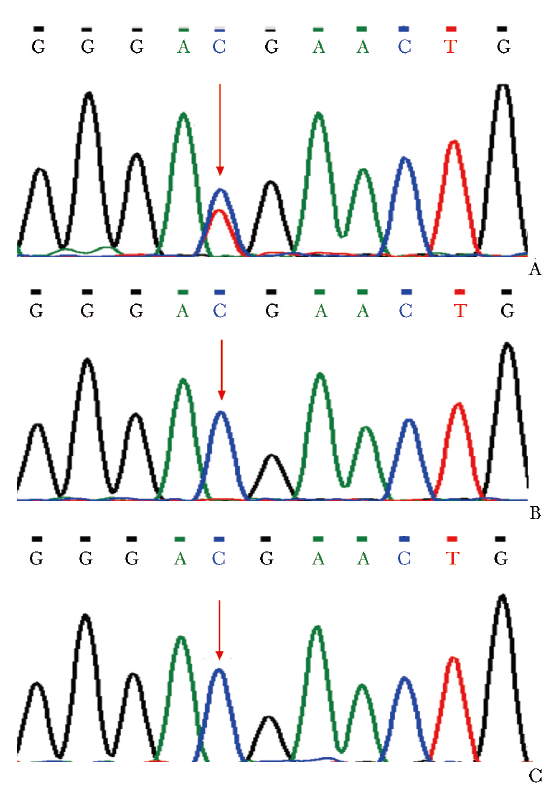
图2 患儿及其父母PTEN基因Sanger测序图示患儿存在PTEN c.388C>T杂合突变(A),患儿父亲(B)和母亲(C)该位点无突变(箭头)
患儿的免疫学指标检测结果为IgA 0.177 g/L↓(参考范围:0.19~1.75 g/L)、IgG 6.040 g/L(参考范围:2.86~16.8 g/L)、IgM 0.752 g/L(参考范围:0.43~1.63 g/L)。
采用流式细胞术对患儿外周血淋巴细胞进行精细免疫分型[4]、对患儿及其父亲单个核细胞(PBMC)中T细胞亚群及衰老、耗竭、活化相关分子表型进行分析、并检测患儿及其父亲外周血T细胞pS6和pAKT的表达水平。
结果显示CD4+终末分化效应记忆T细胞(TEMRA)、CD8+ TEMRA、过渡性B细胞比例及绝对数均增加,浆母细胞比例正常但绝对数增加,γδT细胞比例及绝对数均减少(表1)。
表1 患儿精细免疫分型结果(×109/L)

患儿外周血CD4+T/CD8+T细胞比例正常(1.740),CD4+T细胞中调节性T细胞(Treg)占比0.069(参考范围:0.050~0.100),滤泡辅助性T细胞(Tfh)比例正常(0.163/0.187),T细胞衰老指标CD57在CD3+CD8+ T细胞中高表达,在CD3+CD4+ T细胞中表达水平大致正常(图3)。
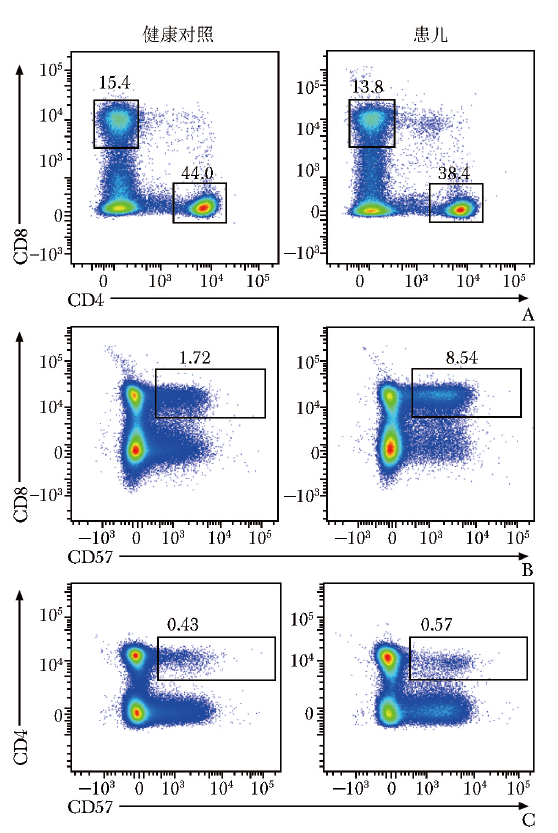
图3 流式细胞术检测患儿T细胞表面CD57的表达情况
A.CD3+CD8+T细胞和CD3+CD4+T细胞比例正常;B.CD3+CD8+CD57+T细胞比例增加;C.CD3+CD4+CD57+T细胞比例正常
患儿T细胞耗竭指标(PD-1)、静息指标(CD62L)及活化指标(CD69、HLADR)数值与健康对照相当,分别为CD3+CD4+PD-1+ T细胞(0.029/0.026),CD3+CD8+PD-1+ T细胞(0.023/0.036)、CD3+CD4+CD62L+T细胞(0.667/0.576)、CD3+CD8+CD62L+T细胞(0.326/0.368)、CD3+CD4+CD69+T细胞(0.632/0.579)、CD3+CD8+CD69+T细胞(0.588/0.565)、CD3+CD4+HLADR+T细胞(0.007/0.004)、CD3+CD8+HLADR+T细胞(0.004/0.004)。患儿外周血T细胞pS6(Ser235/236)和pAkt(Ser473)的平均荧光强度(MFI)在T细胞激活后5 min、15 min、30 min均无明显升高(图4),表明该患儿外周血T细胞PI3K/Akt/mTOR通路磷酸化水平正常。

图4 患儿外周血T细胞PI3K/Akt/mTOR通路磷酸化水平检测
A.pS6(Ser235/236)平均荧光强度;B.pAkt(Ser473)平均荧光强度
采用蛋白质印迹法检测患儿及其父亲PTEN蛋白表达水平,结果显示患儿外周血单个核细胞中PTEN蛋白表达减少(图5)。
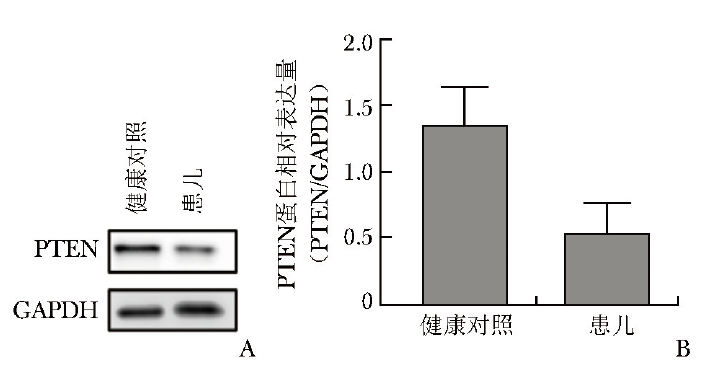
图5 患儿外周血单个核细胞中PTEN蛋白表达水平
A.患儿PTEN蛋白表达减少;B.患儿PTEN蛋白相对表达量降低
2 讨论
Pilarski等[5]于2013年修订了PHTS诊断标准,该标准随后被美国国家综合癌症网络采用[6],符合3条及以上主要标准(但其中1条须为巨头畸形或LDD或胃肠错构瘤)或符合2条主要标准加3条次要标准即可诊断为PHTS。目前本例患儿仅符合2条主要标准(巨头畸形和多发性皮肤黏膜病变)。Busa等[7]认为PTEN突变患儿不同临床表型出现时间具有年龄相关性,考虑到本例患儿年龄尚小,部分表型暂未外显,因此考虑PHTS可能性大。
APDS是由PI3K过度活化引起的原发性免疫缺陷病,能够促进磷脂酰肌醇-4,5-二磷酸(PIP2)磷酸化为磷脂酰肌醇-3,4,5-三磷酸(PIP3),而PTEN恰与PI3K作用相反。PTEN是一种抑癌基因,含9个外显子,位于人10号染色体,其编码的磷脂酰肌醇-3,4,5-三磷酸3-磷酸酶能够促进细胞内PIP3去磷酸化为PIP2,从而拮抗PI3K的作用,实现对PI3K/Akt/mTOR通路的负调控,因此推测PTEN减功能突变(LOF)可导致APDS样表型。
2016年Tsujita等[3]证实PTEN c.697C>T(R233X)杂合突变可导致APDS样表型。APDS患儿常有淋巴增殖表现,如肝脾肿大、淋巴结肿大、结节样淋巴滤泡增生等,伴反复呼吸道感染、支气管扩张、炎症性肠病、自身免疫性疾病、巨细胞病毒和EB病毒易感等[8],本例患儿临床表型与APDS患儿不同,仅肝脏稍大,免疫表型仅部分与APDS患儿相似。
患儿IgM及T细胞PI3K/Akt/mTOR通路磷酸化水平均正常,T细胞耗竭、静息、活化相关指标均正常表达,Treg、Tfh比例正常,而APDS患儿多表现为高IgM,T细胞PI3K/Akt/mTOR通路高度磷酸化,T细胞静息减少,耗竭、活化增加,Treg减少、Tfh增加。
CD57可抑制增殖,是细胞衰老的主要标志物,其主要在TEMRA中高表达。TEMRA是处于分化晚期的T细胞,也可作为衰老标志物[9]。患儿CD4+ TEMRA、CD8+ TEMRA增多,CD8+T细胞表面CD57表达增加,均提示T细胞衰老加速,处于失稳状态,可能原因为IL-7和IL-15受体表达受到抑制[10]。
过渡性B细胞是成熟和未成熟B细胞之间的纽带,在一些自身免疫性疾病(如系统性红斑狼疮)患者中可观察到过渡性B细胞增加,本例患儿过渡性B细胞虽增加但暂无自身免疫相关表现[11]。
PTEN突变患者的临床表型差异较大,目前尚未发现可靠的基因型与表型之间的相关性[12]。PTEN在固有免疫和适应性免疫稳态维持及活化中均有重要作用,理论上均可出现免疫缺陷[13]。
Tsujita等[3]报道了2例携带PTEN LOF突变(p.R233X,p.R15fs)表现为APDS的患者,以及2例同为PTEN LOF突变携带者却无免疫缺陷表现的患者(p.I5fs),上述4例患者T、B细胞PI3K/Akt/mTOR通路磷酸化水平均升高。既往研究报道p.R233X突变患者亦可仅表现为CS[14]。
因此,PTEN LOF突变不一定导致免疫缺陷,其PI3K/Akt/mTOR通路亦可正常,或者免疫细胞虽存在PI3K/Akt/mTOR通路过度活化但无免疫缺陷临床表现。
PHTS患者的健康管理较为复杂,通常涉及定期随访、恶性肿瘤筛查及外科手术[15]。美国国家综合癌症网络给出详细建议,如自7岁起每年进行一次甲状腺超声检查,自40岁起每1~2年进行一次泌尿系超声检查等[16]。
鉴于PTEN对PI3K/Akt/mTOR通路的负调控作用,具有靶向治疗前景。如将mTORC1抑制剂雷帕霉素应用于PTEN+/-小鼠,可抑制其子宫内膜增生和肾上腺髓质瘤细胞生长[17],并且在上皮特异性缺失PTEN的小鼠模型中应用雷帕霉素能够提供化学预防、提高生存率和消退多种病变[18]。目前靶向PI3K通路的生物标志物正被研发用于癌症治疗,其中某些生物标志物或可用于PHTS患者的临床治疗[19-20]。
本文总结了国内首例PTEN基因c.388C>T(p.R130X)新生杂合突变患儿的临床表型和免疫特征,有助于丰富临床医生对该疾病的认识,提高诊治水平。
参考文献
[1]Pilarski R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview[J]. Cancers (Basel), 2019, 11: 844.
[2]Celebi JT, Tsou HC, Chen FF, et al. Phenotypic findings of Cowden syndrome and Bannayan-Zonana syndrome in a family associated with a single germline mutation in PTEN[J]. J Med Genet, 1999, 36: 360-364.
[3]Tsujita Y, Mitsui-Sekinaka K, Imai K, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome-like immunodeficiency[J]. J Allergy Clin Immunol, 2016, 138: 1672-1680.
[4]Ding Y, Zhou L, Xia Y, et al. Reference values for peripheral blood lymphocyte subsets of healthy children in China[J]. J Allergy Clin Immunol, 2018, 142: 970-973.
[5]Pilarski R, Burt R, Kohlman W, et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria[J]. J Natl Cancer Inst, 2013, 105: 1607-1616.
[6]Daly MB, Pilarski R, Berry M, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017[J]. J Natl Compr Canc Netw, 2017, 15: 9-20.
[7]Busa T, Milh M, Degardin N, et al. Clinical presentation of PTEN mutations in childhood in the absence of family history of Cowden syndrome[J]. Eur J Paediatr Neurol, 2015, 19: 188-192.
[8]Coulter TI, Chandra A, Bacon CM, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study[J]. J Allergy Clin Immunol, 2017, 139: 597-606.
[9]Larbi A, Fulop T. From “truly naïve” to “exhausted senescent” T cells: when markers predict functionality[J]. Cytometry A, 2014, 85: 25-35.
[10]Hand TW, Cui W, Jung YW, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival[J]. Proc Natl Acad Sci USA, 2010, 107: 16601-16606.
[11]Zhou Y, Zhang Y, Han J, et al. Transitional B cells involved in autoimmunity and their impact on neuroimmunological diseases[J]. J Transl Med, 2020, 18: 131.
[12]Leslie NR, Longy M. Inherited PTEN mutations and the prediction of phenotype[J]. Semin Cell Dev Biol, 2016, 52: 30-38.
[13]Chen L, Guo D. The functions of tumor suppressor PTEN in innate and adaptive immunity[J]. Cell Mol Immunol, 2017, 14: 581-589.
[14]Nelen MR, van Staveren WC, Peeters EA, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease[J]. Hum Mol Genet, 1997, 6: 1383-1387.
[15]Mester J, Eng C. Cowden syndrome: recognizing and managing a not-so-rare hereditary cancer syndrome[J]. J Surg Oncol, 2015, 111: 125-130.
[16]Daly MB, Pilarski R, Yurgelun MB, et al. NCCN Guide-lines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020[J]. J Natl Compr Canc Netw, 2020, 18: 380-391.
[17]Podsypanina K, Lee RT, Politis C, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/- mice[J]. Proc Natl Acad Sci USA, 2001, 98: 10320-10325.
[18]Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden's disease by mTOR inhibition with rapamycin[J]. Cancer Res, 2008, 68: 7066-7072.
[19]Ross-Innes CS, Becq J, Warren A, et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett's esophagus and esophageal adenocarcinoma[J]. Nat Genet, 2015, 47: 1038-1046.
[20]Lagergren J, Lagergren P. Recent developments in esophageal adenocarcinoma[J]. CA Cancer J Clin, 2013, 63: 232-248.

