约翰·霍普金斯大学Nature发文揭示全球多样化人群中基因表达变异的来源
时间:2024-07-30 14:03:05 热度:37.1℃ 作者:网络
导读
调节基因表达和剪接的遗传变异是影响表型多样性的关键,通过将基因表达和剪接模式与基因组水平的变异相关联,已有研究揭示了这些分子性状的遗传基础及其与高阶表型的关系。虽然这些研究非常有价值,但前期的系列研究多是针对欧洲血统的参与者,因此限制了研究结果的通用性,并阻碍了对全球人类基因表达多样性和进化的理解。
此外,虽然部分研究纳入了不同地理个体的样本,并揭示了大量与基因表达水平相关的遗传变异,但由于研究通常以小样本量或使用微阵列检测基因表达为特征,这限制了数量性状基因座(QTL)定位的统计能力和分辨率,并阻碍了与现代测序数据集的整合和比较。
近日,美国约翰·霍普金斯大学研究团队在Nature发表了题为“Sources of gene expression variation in a globally diverse human cohort”的文章,报道了新搭建的MAGE,这是一个开放获取的淋巴母细胞样细胞系(LCLs)RNA-seq数据集,包含来自千人基因组计划731个个体的数据组成,这些个体来自五大洲的二十多个群体。研究团队绘制了遗传变异与附近基因(分别为顺式表达数量性状位点(eQTL)和顺式剪接QTL(sQTL))表达和剪接之间的关联,并鉴定了超过15000个推测的因果eQTL和超过16000个因果sQTL,这些QTL富集了相关的表观基因组特征。此外,因果eQTL效应的大小和方向在不同群体中高度一致。总之,该研究扩展了对人类基因表达多样性的理解,并为研究人类基因组的进化和功能提供了一个重要资源。

文章发表在Nature
主要研究内容
基因表达和剪接多样性
首先,研究人员对来自千人基因组计划的731个个体进行RNA-seq,代表26个全球分布的种群(每个种群27-30个个体),跨越5个大洲。研究使用GENCODE(v.38)的基因注释定量基因表达水平,使用LeafCutter定量可变剪接模式。整体而言,有分别超过92%和95%的表达和剪接变化发生在群体内而不是群体之间,说明基因对转录组性状的影响在不同的血统群体中是相对一致的,而群体之间的表达水平变化大于剪接变化。
值得注意的是,研究团队还观察到表达和剪接的群体内方差在大陆群体之间存在差异;与混合的美洲大陆组相比,在非洲大陆组内观察到了更高的平均方差,这些结果与人类全球迁徙期间遗传多样性下降一致。
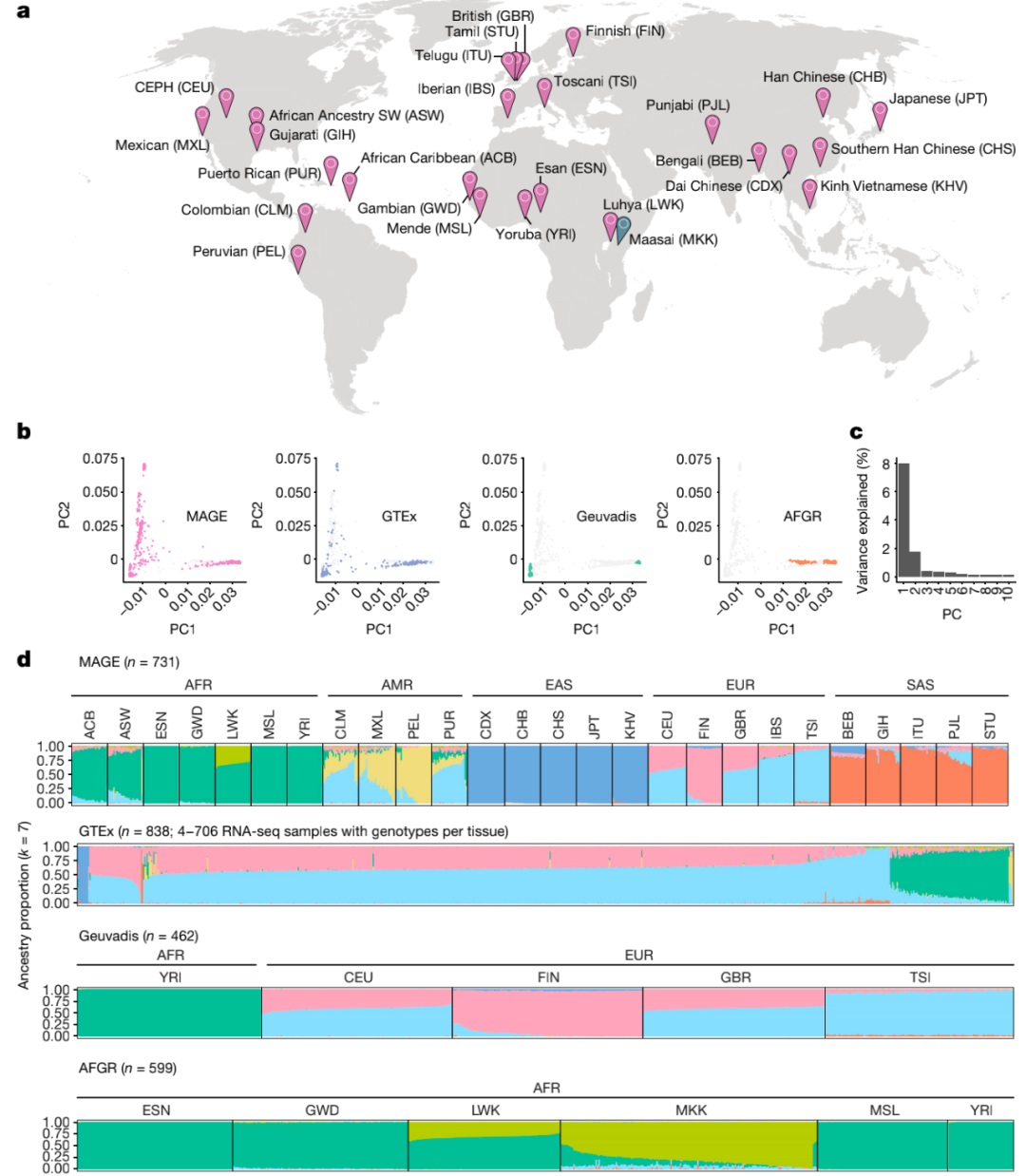
图1. 全球多样性转录组学数据集的构建
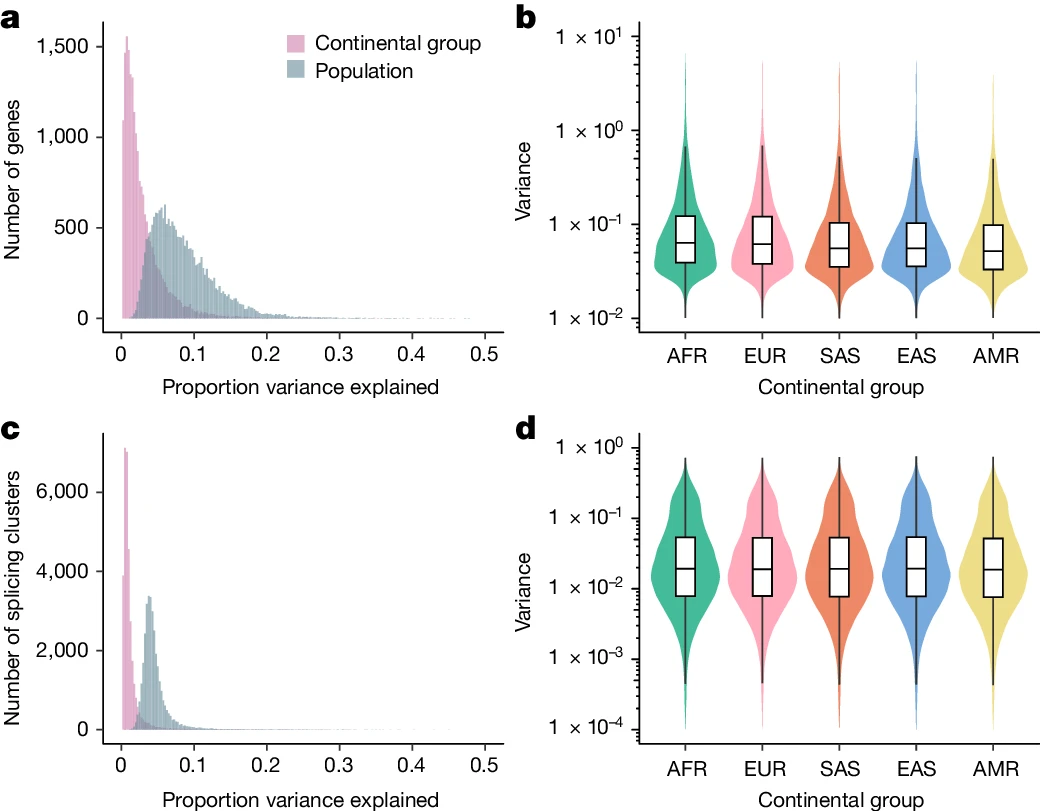
图2.群体内和群体间转录组多样性的模式。
高分辨率绘制eQTLs和sQTLs
MAGE能够揭示驱动基因表达和剪接变化的遗传因素。通过交叉来自同一组样本的已发表的基因型数据,研究人员在每个基因的转录起始位点(TSS)的1Mb内定位了顺式eQTL和顺式sQTL,并将eGenes和sGenes分别定义为具有eQTL或sQTL的基因,将eVariants和sVariants分别定义为具有eQTL或sQTL信号的个体遗传变体。
此外,研究人员使用SuSiE对所有eGenes和所有sGenes的内含子进行精细定位,以识别驱动每个QTL信号的因果变体。对于每个基因和内含子,SuSiE鉴定一个或多个可信集合,代表独立的因果eQTL和sQTL信号。研究观察到精细定位基因之间广泛的等位基因异质性,其中40%精细定位的eGenes和53%精细定位的sGenes显示出一个以上的不同可信集。研究还在识别驱动表达变化的推定因果变异方面实现了高分辨率,在15664个eQTL可信集合中,3992个包含单个变异;类似地,对于sQTL,16451个可信 中的3569个包含单个变异。
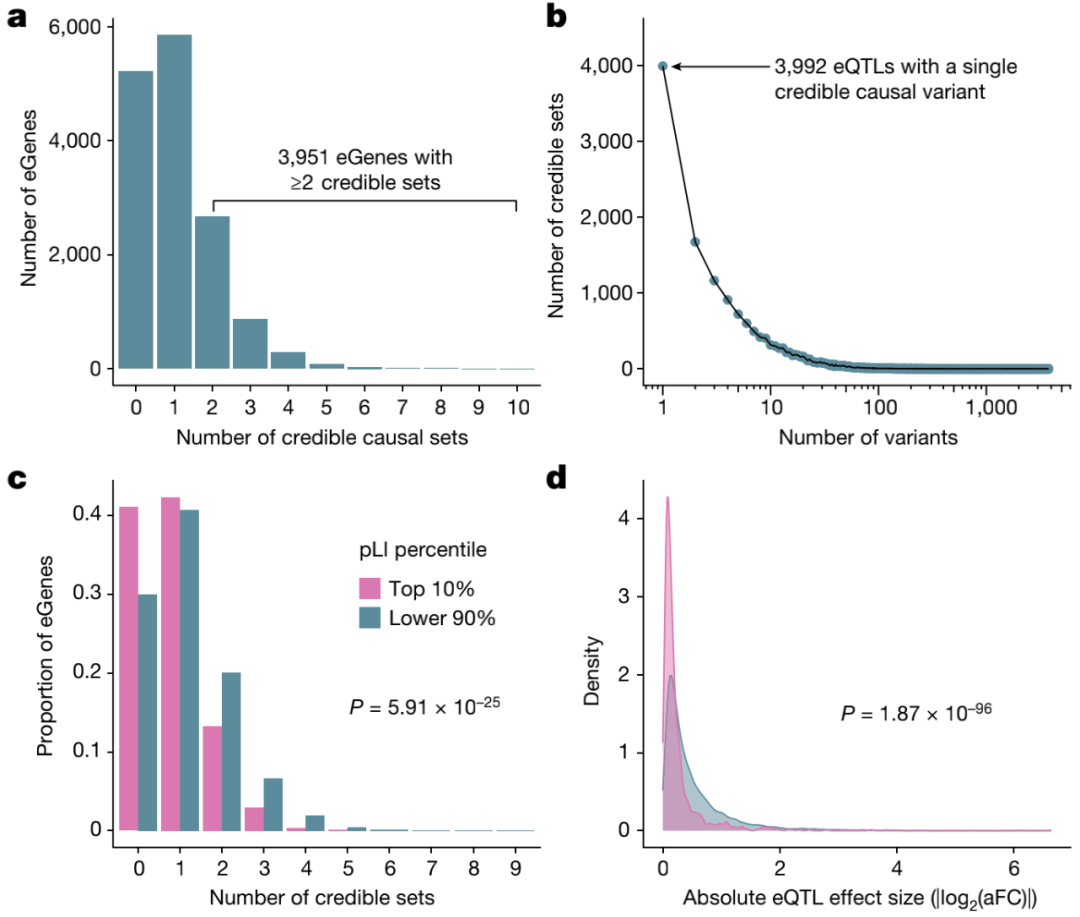
图3.高分辨率eQTL的定位
QTL的功能富集
利用推定的因果信号,研究团队量化了来自Roadmap Epigenomics chromHMM模型中127个参考表观基因组中注释染色质状态的精细定位的前导eQTL富集。结果发现,富集在启动子区域内最明显,特别是在活性TSS(TssA)和侧翼区域(TssAFlnk),增强子区域(Enh和EnhG)内的富集也相对明显,尤其是对于血细胞类型来说。
对表观基因组富集和eQTL效应大小间的关系进行分析发现,启动子相关富集在eQTL效应大小的十分位数中是一致的,并且在平衡调控区域内的富集对于效应较大的eQTL最为明显;相比之下,位于与转录区相关的染色质状态内的eQTL主要表现出较低的效应量。此外,相对于增强子和同时充当启动子和增强子的区域,启动子区域中拥有更大的中值eQTL效应。总的来说,以上发现为精确定位的顺式QTL的生物学有效性提供了支持。
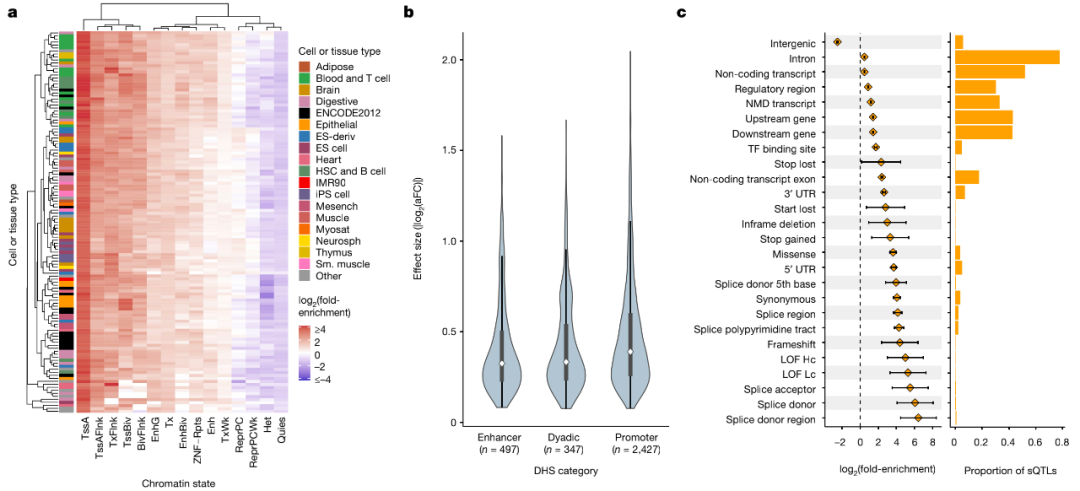
图4. 顺式QTL在多种细胞和组织类型调控区域中的富集分析
eQTLs、sQTLs和GWAS的共定位
为了更好理解遗传变异与生物表型联系起来的分子机制,研究团队将eQTL和sQTL与全基因组关联分析(GWAS)数据进行交叉分析,并从中鉴定了54个与eQTL和sQTL信号共定位的GWAS信号。QTL和GWAS之间的有限共定位主要源于形成遗传变异的不同选择压力。GWAS分析倾向于发生在纯化选择下的基因内,而分子QTL则在放松限制下最容易识别。
进一步的分析数据表明,因果eQTL效应的大小和方向在不同群体中高度一致。需要注意的是,在前期研究中观察到的明显的“群体特异性”效应主要是由未检测到的相同基因的低分辨率或额外的独立eQTL驱动的。
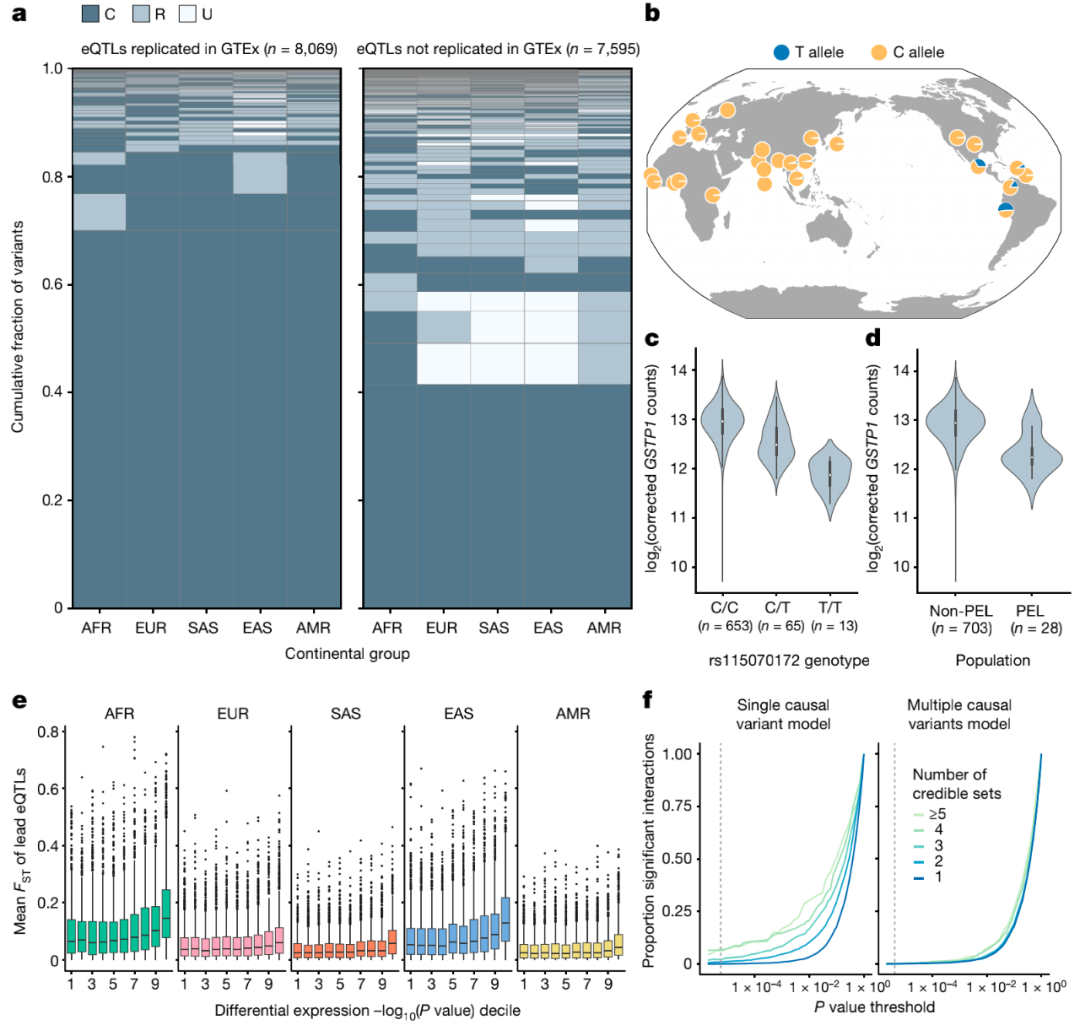
图5. 基因表达的群体特异性遗传效应
结 语
综上所述,该研究结合现有全基因组测序数据和新的转录组测序数据,提供了一个大型开放获取的数据集MAGE,可用于研究多祖先人类基因表达和剪接的多样性和进化。该研究还提供了深入了解这些关键分子表型变异的遗传来源。在现有的高覆盖率全基因组测序数据的补充下,预计MEGA将成为一个宝贵的数据资源,以促进未来对人类基因组功能变异的复杂遗传基础的研究。
文章通讯作者Rajiv C. McCoy表示:“我们找出了成千上万影响基因表达和剪接模式的基因组变异,这些变异可能反过来影响人体的各种性状。其中一部分变异,是在以前研究里代表性不足的人群里才有的。我们希望该结果能帮助减少不同血统群体之间的一些历史健康差距,同时,该研究是为了得出更普遍相关和能用的结果。我们正在走进个体化精准医疗的时代,希望该研究能给更公平的医疗进步奠定基础。”
论文原文:
Taylor, D.J., Chhetri, S.B., Tassia, M.G. et al. Sources of gene expression variation in a globally diverse human cohort. Nature (2024). https://doi.org/10.1038/s41586-024-07708-2

