罕见病专栏|中年男性,进行性行走困难——遗传性痉挛性截瘫7型二家系
时间:2025-02-27 12:36:35 热度:37.1℃ 作者:网络
摘 要 本文报告2例由SPG7基因突变所致遗传性痉挛性截瘫7型(spastic paraplegia, SPG7)病例。2例患者均为中年男性,表现为成年起病的进行性加重的痉挛性步态、构音障碍及共济失调。此外,家系1先证者合并有二便功能障碍。家系2先证者还伴有癫痫发作,其辅助检查提示小脑轻度萎缩,并伴有部分感觉神经受损和肌肉的神经源性损害。全外显子测序提示二人均由SPG7复合杂合突变所致。目前,本病以对症支持治疗为主。通过对相关文献进行回顾,总结了本病的基因型和表型特点,以期进一步提高对该病的认识,并为临床诊治提供参考。
关键词
遗传性痉挛性截瘫7型;SPG7;共济失调;小脑萎缩;癫痫
1 临床资料
1.1 家系1 先证者(Ⅱ-2,图1A),男,40岁,因“双下肢僵硬行走困难8年”就诊。患者自32岁起出现进行性走路困难,易摔倒,不能跑步。近两年症状明显加重,上下楼梯需搀扶,偶有饮水呛咳,但无吞咽困难,并伴有大便失禁,偶有小便失禁。患者既往有腰椎间盘突出和副鼻窦炎病史,否认外伤或其他慢性病史,否认类似症状家族史。
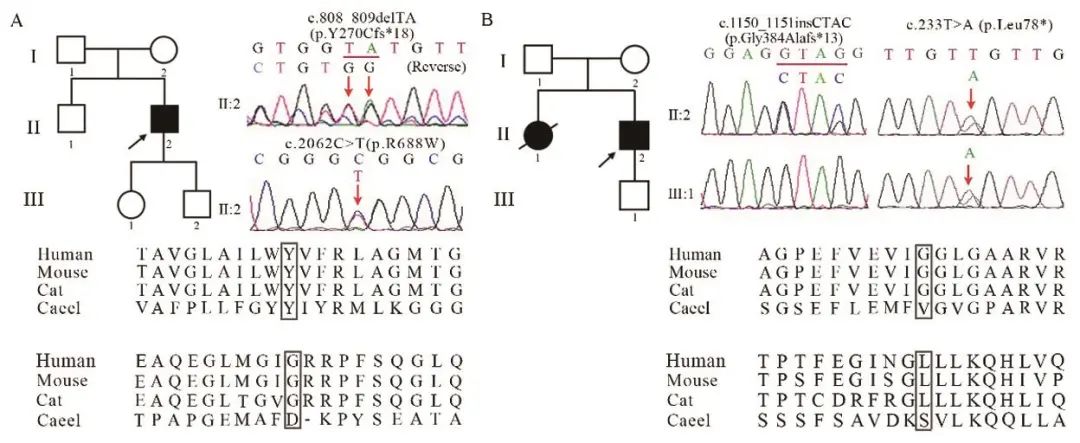
图1 SPG7患者家系图、保守性分析及基因突变示意图 A:家系1的家系图列于左上,箭头所示为先证者;Ⅱ-2一代测序图示SPG7基因复合杂合突变(c.808_809delTA和c.2062C>T);突变的氨基酸保守性分析列于下方。B:家系2的家系图列于左上,箭头所示为先证者;Ⅱ-2一代测序图示SPG7基因复合杂合突变(c.1150_1151insCTAC和c.233T>A);Ⅲ-1为c.233T>A的杂合携带者;突变的氨基酸保守性分析列于下方。
体格检查:神清,吐词欠流利,双眼水平细微眼震,无肌肉萎缩或疼痛,双下肢肌张力增高,肌力5级,轮替试验差,一字步不能,剪刀步态,四肢深浅感觉对称无异常。双下肢腱反射亢进,双侧Babinski征阳性,踝阵挛阳性(表1)。
辅助检查:头颅磁共振未见异常;肌电图正常,血常规红细胞5.8×1012/L[(4.0~5.5)×1012/L],血红蛋白161 g/L(120~160 g/L);叶酸12.10 nmol/L(11~54 nmol/L)。
基因检查:抽取家系1中的患者Ⅱ-2肘静脉外周血,进行全外显子测序(whole exome sequencing, WES),结果显示SPG7(spastic paraplegia 7)基因存在复合杂合突变:c.808_809delTA(p.Y270fs*18)和c.2062C>T(p.R688W)(图1A),分别来自父亲和母亲。其中c.2062C>T(p.R688W)为已知突变,c.808_809delTA(p.Y270fs*18)为本文新报告的突变。根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics, ACMG)指南[1],上述突变均被评为“致病”或“可能致病”。SPG7基因(NM_003119)测序图及突变的氨基酸序列保守性分析见图1A。
1.2 家系2 先证者(Ⅱ-2,图1B),男,50岁,因“进行性双下肢僵硬行走困难3年,3个月内反复意识丧失3次”就诊。患者自47岁起无明显诱因下出现双下肢僵硬,行走不稳,伴有双足底麻木感,并于行走后加重,休息时无明显缓解。2年前出现口齿不清,不影响交流,无饮水呛咳及吞咽困难,无肌肉萎缩及肌肉疼痛。3个月前,在床上休息时突然出现意识丧失,呼之不应,双眼直视,伴有小便失禁,无牙关紧闭,无明显肢体抽搐,持续约30 min意识转清;近2个月内又先后发生2次意识丧失,每次持续2~3 min,此后未再发作,就诊于当地医院诊断为“癫痫”。既往有腰椎间盘突出和双侧上颌窦、筛窦及额窦炎病史,否认外伤传染病史,姐姐(II-1)生前有类似症状,具体死因不明。
体格检查:神清,构音含糊,双眼一过性水平眼震,双下肢肌张力增高,肌力5级,指鼻试验、跟膝胫试验稳准,闭目难立征可疑阳性,剪刀步态,四肢深浅感觉对称无异常,无肌肉萎缩或疼痛。咽反射减弱,双侧膝反射活跃,双侧病理征未引出,踝阵挛阳性。
辅助检查:查颈椎MRI示C3~6椎间盘突出;腰椎MRI示L3~5椎间盘突出;头颅MRI示小脑轻度萎缩。双下肢肌电图:左腓总运动神经及右腓浅感觉神经受损;左胫前肌、右股四头肌神经源性损害(表1)。
基因检查:抽取家系2中的先证者Ⅱ-2肘静脉外周血进行全外显子测序,发现SPG7基因存在复合杂合突变c.1150_1151insCTAC(p.Gly384Alafs*13)以及c.233T>A(p.Leu78*)(图1B)。Ⅱ-2除了具有皮质脊髓束损害的临床特点外,还有反复癫痫发作病史。家族中Ⅱ-1生前也存在相似临床症状,已去世未获得相应组织标本。Ⅲ-1为c.233T>A(p.Leu78*)的携带者(图1B)。SPG7基因(NM_003119)测序图及突变的氨基酸保守序列信息详见图1B。
2 讨论
遗传性痉挛性截瘫(spastic paraplegia,SPG)是一种遗传性皮质脊髓束运动神经系统退行性疾病,表现为下肢的进行性痉挛和无力,已知的基因亚型已超过80种[2]。其中SPG7亚型,约占AR-HSP(autosomal recessive hereditary spastic paraplegias)患者的5%[3]。单纯型SPG7表现为双下肢进行性痉挛性瘫痪。复杂型SPG7除具有上述特点外,还伴有视神经受累、皮质和小脑萎缩、肌肉萎缩、智力发育迟缓、构音障碍、周围神经病变等[4],部分病例合并癫痫发作、帕金森样症状等[5]。SPG7在全球人群发病风险为1:100 000~9:100 000[3]。大多数患者为成年起病(发病年龄11~72岁)[6]。
SPG7基因位于 16q24.3[7],包含有17个外显子[8]。截止目前,我国既往已报告突变30种。全球已报告的与HSP相关的SPG7基因突变有200余种(总结见图2)[9-26],其中点突变124种(61.7%),缺失突变34种(16.9%),剪切突变26种(12.9%),重复突变11种(5.5%),插入突变4种(2.0%),插入缺失1种(1.0%)。最常见的致病性突变是位于11号外显子的c.1529 C>T(p.Ala510Val),并以11、13和15号外显子为热点突变区域[27]。
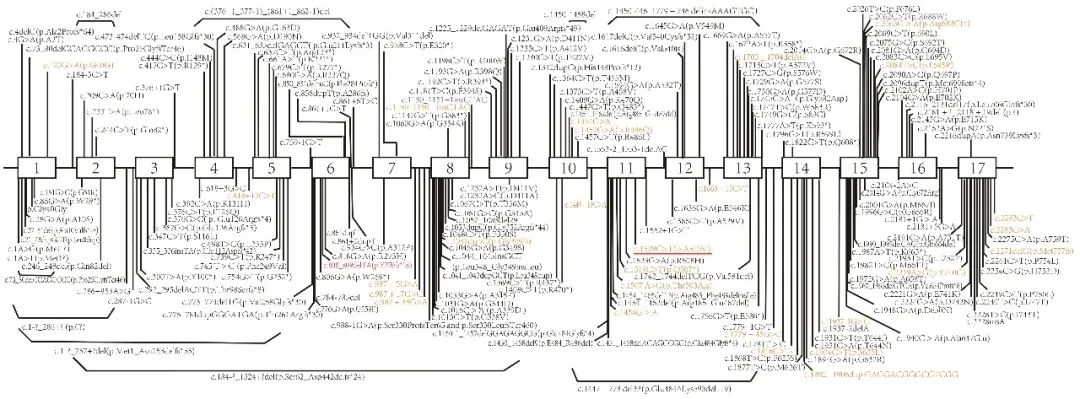
图2 SPG7患者家系图、保守性分析及基因突变示意图 SPG7基因共包含17个外显子,全球已报告突变如图所示。橙色:我国已报告突变。红色:本文新发先突变。蓝色:既往报告与本文突变重叠的位点。红色下划线:热点突变。
SPG7以常染色体隐性遗传为主,少数报告称本病可呈常染色体显性遗传[7, 21, 28-29],但这些报告通常为个案,且多未经可靠家系共分离验证[2],因此对于SPG7亚型是否存在常染色体显性遗传方式存在争议[28]。小脑性共济失调是SPG7的重要临床特点之一[30]。家系1先证者除具有典型的HSP表现外,还合并有共济失调和二便障碍;家系2先证者除典型HSP表现外,还伴有癫痫发作、周围神经损害、共济失调体征,因此两例均为复杂表型。在西班牙的HSP临床研究中,c.233T>A(p.Leu78*)杂合突变曾被报告可能与SPG7单纯型表型有关;但在意大利的两例相同突变杂合携带者中并无临床症状[7]。本研究家系2中,先证者的儿子为c.233T>A(p.Leu78*)的杂合携带者,目前亦无临床症状。
SPG7基因在不同组织和不同生长发育阶段的表达水平各异,在人类中枢神经系统的杏仁核、尾状核、丘脑等部位高表达,而在黑质等部位呈低表达[20]。该基因编码的产物是Paraplegin蛋白[31],定位于线粒体内膜,在调控细胞降解错误折叠蛋白和核糖体生成中具有重要作用[32]。Paraplegin缺陷导致线粒体内ATP合酶和呼吸链复合体合成障碍,致使线粒体功能失调,并引起轴突退行性改变[33]。
SPG7突变不仅可以导致遗传性痉挛性截瘫,还可导致肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)、原发性侧索硬化(primary lateral sclerosis,PLS)、腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT)、脊髓小脑共济失调(spinocereballar ataxia,SCA)、髓鞘发育不良性脑白质营养不良(hypomyelinating-leukodystrophy disorders,HLD)和多系统萎缩(multiple system atrophy, MSA)等表型[34]。此外,SPG7基因突变相关的HSP具有高度临床异质性[35],如共济失调、构音障碍、括约肌功能障碍、痉挛和帕金森病等[11, 36-37];部分SPG7突变与小脑性共济失调相关[10],例如携带至少一个p.Ala510Val突变的患者,更易出现小脑性共济失调,发病年龄更迟[11]。但目前全球尚无确切的基因型-表型相关性研究证据[38]。本文报告的两个家系均伴有共济失调表现,这是SPG7相比于其他亚型较为特殊的临床表现之一[39]。
本文共检出四种SPG7基因突变c.808_809delTA(p.Y270fs*18)、c.2062C>T(p.R688W)、c.1150_1151insCTAC (p.Gly384Alafs*13)以及c.233T>A(p.Leu78*),其中c.808_809delTA(p.Y270fs*18)为新报告突变;c.1150_1151insCTAC和c.2062C>T(p.Arg688Trp)在我国SPG7患者中已被报告[40];c.233T>A(p.Leu78*)位于SPG7基因2号外显子,曾于欧洲等地被报告[7](图1C)。
临床工作中,对于伴有共济失调的HSP患者,应首先筛查SPG7基因突变。与此同时,还应注意与其他以小脑性共济失调为主要临床表现的疾病相鉴别,例如 SCA和MSA-C等[41]。本病尚无有效治疗方法,以对症支持和康复训练为主。但已有研究使用基因治疗方法对其他HSP亚型进行干预,并在动物模型上取得了一定进展,可能为未来本病的治疗性研究提供借鉴意义[42]。
3 点评
SPG7呈常染色体隐性遗传,是AR-HSP的常见类型之一。SPG7基因突变的形式包括移码突变、错义突变和缺失突变等。随着更多病例的表型与基因型特点被报道,对该疾病的认识与研究也在不断深入,其临床症状复杂多样性及发病机制之谜将被解开,同时也将为其他潜在的治疗靶点的研究提供新的视野。
参考文献:
1. SUE R, NAZNEEN A, SHERRI B, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5): 405-424.
2. MEHRDAD A E, ERIC Y, IKHLASS H S, et al.Evidence for Non-Mendelian Inheritance in Spastic Paraplegia 7[J]. Mov Disord, 2021, 36(7): 1664-1675.
3. ERIC G. Contributions of the basal ganglia to action sequence learning and performance[J]. Neurosci Biobehav Rev, 2019, 107: 279-295.
4. GAUTAM W, KISHORE R K, ERANDHI L, et al.Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells[J]. Front Neurosci, 2020, 14: 820.
5. CHRISTOPHER J M, CHRISTINE E B, JANINE K, et al. Clinical features of hereditary spastic paraplegia due to spastin mutation[J]. Neurology, 2006, 67(1): 45-51.
6. JHNATHAN D M, SHARON T, JONATHAN D F, et al. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature[J]. Acta Neuropathol, 2004, 107(6): 481-488.
7. SANCHEZ-FERRERO E, COTO E, BEETZ C, et al.SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p.A510V[J]. Clin Genet, 2013, 83(3): 257-262.
8. IZADORA FONSECA Z S, VINICIUS BOARATTI C, DAVID F, et al. Cognitive dysfunction and psychosis: expanding the phenotype of SPG7[J]. Neurocase, 2021, 27(3): 253-258.
9. BEATRIZ DE LA C F, GORKA F E, JOSEP G, et al. Parkinsonism and spastic paraplegia type 7: Expanding the spectrum of mitochondrial Parkinsonism[J]. Mov Disord, 2019, 34(10): 1547-1561.
10. CECILIA M, ELISA G, ANNA R, et al. Prevalence and phenotype of the c.1529C>T SPG7 variant in adult-onset cerebellar ataxia in Italy[J]. Eur J Neurol, 2019, 26(1): 80-86.
11. GIULIA C, REBECCA S, BART P.C V D W,et al. Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7[J]. Neurology, 2019, 92(23): e2679-e2690.
12. CE K, CHRISTINA L, KATE E A, et al. High Degree of Genetic Heterogeneity for Hereditary Cerebellar Ataxias in Australia[J]. Cerebellum, 2019, 18(1): 137-146.
13. MARINA C R, RAQUEL B M, ISABEL S B, et al. Hereditary Spastic Paraplegia 7 Presenting as Multifocal Dystonia with Prominent Cranio-Cervical Involvement[J]. Mov Disord Clin Pract, 2021, 8(6): 966-968.
14. DANIELE G, GIOVANNA D M, GABRIELLA S, et al. NGS in Hereditary Ataxia: When Rare Becomes Frequent[J]. Int J Mol Sci, 2021, 22(16): 8490.
15. DANIELA B, MARCIA P B, LAIS ALVES JACINTO S, et al. Clinical and molecular characterization of hereditary spastic paraplegias: A next-generation sequencing panel approach[J]. J Neurol Sci, 2017, 383: 18-25.
16. PETYA B M, HONGYING C, HELENA MARIA P, et al. Neurophysiological and ophthalmological findings of SPG7-related spastic ataxia: a phenotype study in an Irish cohort[J]. J Neurol, 2021, 268(10): 3897-3907.
17. ISELIN MARIE W, JEANETTE K, GIA TUONG T, et al. Spastic paraplegia type 7 is associated with multiple mitochondrial DNA deletions[J]. PLoS One, 2014, 9(1): e86340.
18. HUSSEIN D, ELENI MERKOURI P, BOUCHRA O A B, et al. Identification of a novel homozygous SPG7 mutation by whole exome sequencing in a Greek family with a complicated form of hereditary spastic paraplegia[J]. Eur J Med Genet, 2015, 58(11): 573-577.
19. SCHLIPF N A, SCHULE R, KLIMPE S, et al. Amplicon-based high-throughput pooled sequencing identifies mutations in CYP7B1 and SPG7 in sporadic spastic paraplegia patients[J]. Clin Genet, 2011, 80(2): 148-160.
20. CHATRI S, SCOTT A W, JOANNA C, et al. Genomic structure and expression analysis of the spastic paraplegia gene, SPG7[J]. Hum Genet, 1999, 105(1-2): 139-144.
21. PHILIP A W, ANDREW H C, CHRISTOPHER T, et al. A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia[J]. Brain, 2004, 127(Pt 5): 973-980.
22. MARIOS H, JOHN M, PRIYA S, et al. Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients[J]. J Neurol Neurosurg Psychiatry, 2017, 88(4): 301-309.
23. YI Y, LEI Z, DAVID R L, et al. Compound heterozygote mutations in SPG7 in a family with adult-onset primary lateral sclerosis[J]. Neurol Genet, 2016, 2(2): e60.
24. YIHUI L, JJIANG X, WANYUN T, et al. Exome Sequencing Identifies a Mutation (Y740C) in Spastic Paraplegia 7 Gene Associated with Adult-Onset Primary Lateral Sclerosis in a Chinese Family[J]. Eur Neurol, 2019, 81(1-2): 87-93.
25. SHAKYA B, VICTORIA S, DIANE C, et al. Novel c.775_781dup, p(Thr261fs) mutation in SPG7 gene in a patient with hereditary spastic paraparesis[J]. Neurol India, 2017, 65(5): 1141-1142.
26. CHANNA A H, NIGEL H, RONAN O M, et al. Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations[J]. Neurology Genetics, 2018, 4(6): e279-e289.
27. LINWEI Z, KAREN N M, SANKARASUBRAMON H S, et al. SPG7 and Impaired Emotional Communication[J]. Cerebellum, 2017, 16(2): 595-598.
28. ESGARD V, AGATHA S, GORKA F E, et al. A deep intronic splice variant advises reexamination of presumably dominant SPG7 Cases[J]. Ann Clin Transl Neurol, 2020, 7(1): 105-111.
29. STEOHAN K, CHRISTEL D, SYLVIE G, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy[J]. Basal Ganglia, 2013, 3(1): 69.
30. MATTHIS S, REBECCA S. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways[J]. Mov Disord, 2017, 32(3): 332-345.
31. FATIMA F, ANGELO Q, MARINELLA P, et al.Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport[J]. J Clin Invest, 2004, 113(2): 231-242.
32. MIRKO K, METODI D M, GIORGIO C, et al.Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia[J]. Mol Cell Biol, 2007, 27(2): 758-767.
33. GAUTAM P, RUTH E T, LEO J P. Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration[J]. Cell Death Dis, 2018, 9(3): 304.
34. PAULO VICTOR SGOBBI S, THIAGO B, RENAN BRAIDO D, et al.New genetic causes for complex hereditary spastic paraplegia[J]. J Neurol Sci, 2017, 379: 283-292.
35. NICOLAS C, NICOLAS D, ZIV G O, et al.Clinical and genetic study of hereditary spastic paraplegia in Canada[J]. Neurol Genet, 2017, 3(1): e122.
36. KARINE C, MARTINE T, SHARON Y, et al.SPG7 mutations explain a significant proportion of French Canadian spastic ataxia cases[J]. Eur J Hum Genet, 2016, 24(7): 1016-1021.
37. SHAKYA B, MUHAMMAD N, MARTIN S. Early Onset Degenerative Parkinsonism-Consider SPG7 Mutation[J]. Neurol India, 2021, 69(4): 1051-1052.
【引用格式】蒋凯丽,朱泽宇,钟平,等. 中年男性,进行性行走困难——遗传性痉挛性截瘫7型二家系[J]. 中国神经精神疾病杂志,2022,48(5):315-320.
【Cite this article】JIANG K L, ZHU Z Y, ZHONG P, et al. Tow middle-aged males with progressive walking difficulties —— spastic paraplegia type 7 due to SPG7 gene mutations[J]. Chin J Nervous Mental Dis,2022,48(5):315-320.
DOI:10.3969/j.issn.1002-0152.2022.05.013

