JCEM:BDV综合征:一种类似Prader-Willi综合征的严重肥胖和神经发育迟缓的新兴综合征
时间:2021-10-23 12:02:57 热度:37.1℃ 作者:网络

背景:CPE编码羧肽酶E,这是一种将前肽和前肽激素转化为生物活性形式的酶。广泛表达于内分泌和中枢神经系统。到目前为止,已经报道了来自2个家庭的4名患者,他们的主要临床特征包括病态肥胖、神经发育迟缓和促性腺功能减退,并携有双等位基因功能丧失(LoF) CPE变异。
目的:我们描述了来自3个不同血缘家庭的4个受影响的个体,2个叙利亚人的兄弟姐妹,1个埃及人的兄弟姐妹,1个巴基斯坦人的后裔,所有人都有新的纯合子CPE LoF变异。
方法:在排除Prader-Willi综合征(PWS)后,对两名叙利亚同胞进行外显子组测序。在一个大规模的外显子组研究和ClinVar数据库中,在另外2个个体中发现的变异报告为研究变异。所有可能错义变化的计算建模允许评估CPE对错义变体的耐受性。
结果:所有患者均为严重肥胖并伴有神经发育迟缓和其他内分泌异常。来自2个家族的3个个体具有相同的CPE纯合子截断变异c.361C > T, p.(Arg121*),而第4个个体携带c.994del, p.(Ser333Alafs*22)。与之前描述的病例的临床特征进行比较,并根据人类表型本体论术语进行标准化,表明了一种可识别的临床表型,我们称之为Blakemore-DurmazVasileiou (BDV)综合征。计算分析表明CPE结构域的高度保守性和对误义变化的不耐受。
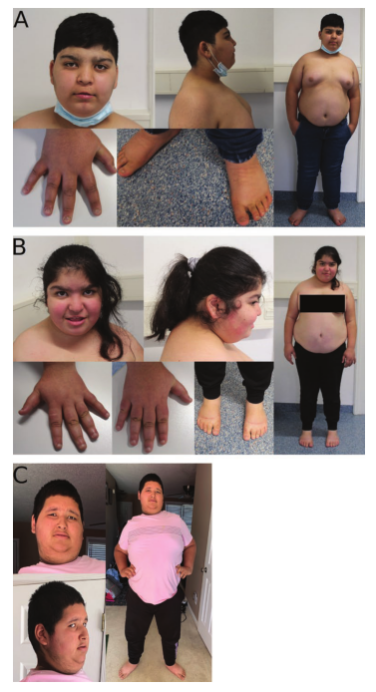
图1 A, 15岁零3个月时C-1个体的面部表型和全身图像;B, 13岁零3个月的妹妹C-2;C, E-1个体在15岁零8个月时。请注意这三个人的严重肥胖。常见的畸形特征包括椭圆形的脸,面部粗糙,异常的睑裂,丘比特弓状上唇,外翻的下唇朱红,小颌,尖细的手指和扁平足。另外,注意兄弟姐妹C-1和C-2的上唇红肿、短指、指甲和脚趾甲发育不良以及个别C-1的指甲和脚趾甲营养不良。
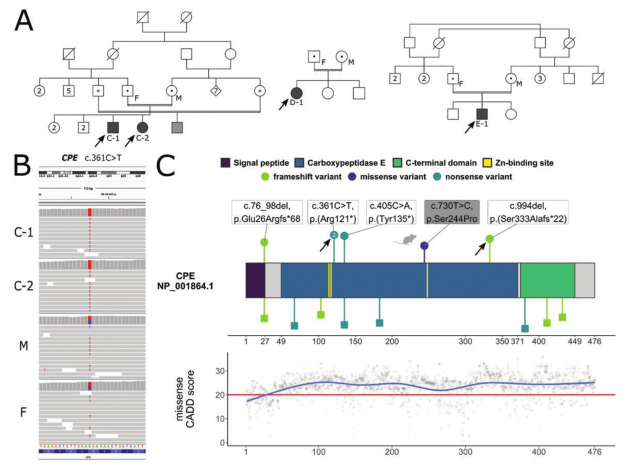
图2 A.本文报告的家系的家系。C-1、C-2、D-1和E-1是索引情况(也由箭头和黑色阴影方框表示)。黑点代表母亲(M)和父亲(F)的杂合子状态。灰色方块表示的是C-1和C-2在叙利亚的表亲,但未经检测,而他的父母和E-1的父亲的白色圆点用黑色轮廓表示假定的杂合性。双线表示血缘关系;B,IGV的代表性图像,显示已识别的C.361C>T变体在C-1和C-2中的位置。注意受影响兄弟姐妹的纯合性和父母的杂合性状态。上图:羧肽酶E(CPE)及其结构域的线性蛋白质结构。CPE由一个N-端信号肽、一个含有3个锌结合位点的中心酶活性结构域和一个可能协助蛋白质折叠、蛋白质-蛋白质相互作用或酶调节的C-末端结构域组成。这里和先前描述的纯合功能丧失(LoF)致病变体在蛋白质模型上方和在蛋白质模型下方的基因组聚集数据库(GnomAD)中注释的所有杂合LoF变体(绿色:移码、紫色:错义、汽油:无义;圆圈:纯合;正方形:杂合;片段长度:比例组合注释依赖耗竭[CADD]得分)表示。箭头表示这里描述的致病变异体c.361C>T,p.(Arg121*)和c.994del,p.(Ser333Alafs*22)。对应的c.361C>T,p.(Arg121*)中的数字2表明,到目前为止,该变体已在2个家系中被报道。在小鼠中已知的、被定位到人类CPE转录本上的错义变体为c.730T>C,p.(Ser244Pro),蛋白质模型上方的灰色标记的老鼠符号也表示该错义变体。下图:所有可能的CPE错义变种的CADD评分的广义线性模型。蓝线表示平滑的CADD值,而红色水平线表示此分数的推荐分界值(另请参阅补充测试表3)(53,54)。总体而言,除N端信号肽外,CPE蛋白高度保守。
结论:双等位基因截尾CPE变异与BDV综合征有关,BDV综合征是一种临床可识别的单基因隐性综合征,伴儿童期肥胖、神经发育迟缓、促性腺功能减退和甲状腺功能减退。BDV综合征类似于PWS。我们的研究结果表明,错义变异也可能与临床相关。
原文出处:
Bosch E, Hebebrand M, Popp B,et al.BDV-syndrome: An emerging syndrome with profound obesity and neurodevelopmental delay resembling Prader-Willi syndrome.J Clin Endocrinol Metab 2021 Aug 12

