阵发性运动障碍(Paroxysmal dyskinesia,PxD)
时间:2023-10-02 14:17:53 热度:37.1℃ 作者:网络
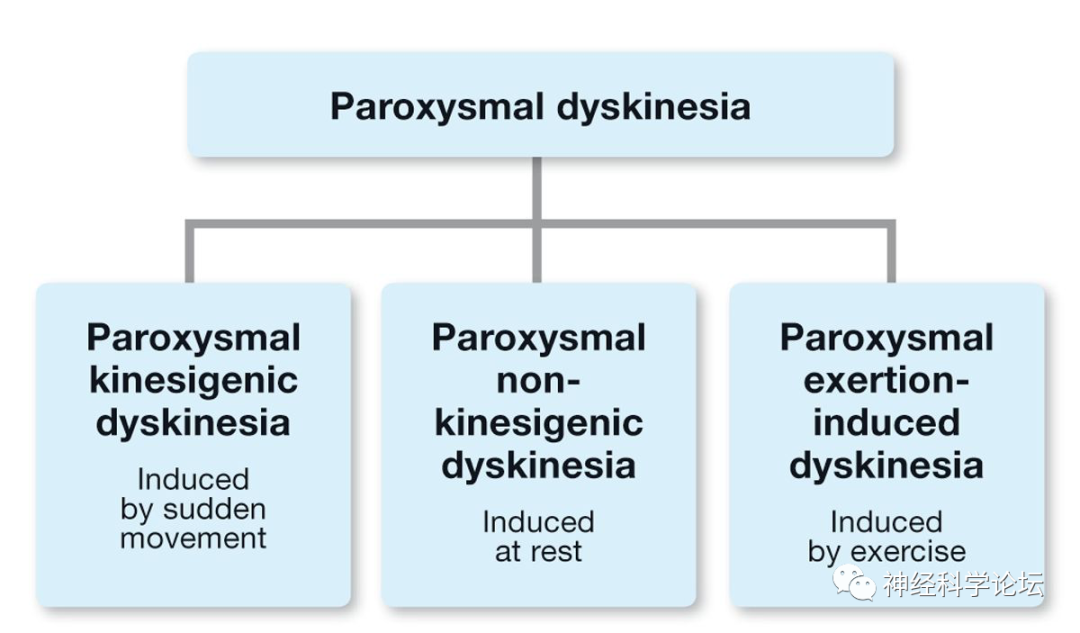
论坛导读:发作性运动障碍疾病(Paroxysmal movement disorders)传统上被分为发作性运动障碍(Paroxysmal dyskinesia,PxD) 和发作性共济失调 (episodic ataxia ,EA )。发作性运动障碍包括无意识丧失的不随意运动发作(主要是肌张力障碍和/或舞蹈病),发作性共济失调的特征是伴有或不伴有发作间神经表现的小脑功能障碍发作。发作性运动障碍疾病涉及离子通道以及与囊泡突触周期相关或与神经元能量代谢有关的蛋白质。阵发性运动障碍(Paroxysmal dyskinesia,PxD)是一组异质性综合征,以可检测因素触发的异常运动反复发作为特征,无意识丧失。根据诱发因素,它们被分为阵发性运动诱发性运动障碍(PKD)、阵发性非运动诱发性运动障碍(PNKD)和阵发性运动诱发性肌张力障碍(PED)。
阵发性运动障碍(Paroxysmal dyskinesia,PxD)是一种罕见的运动障碍,影响成人和儿童。基于诱发异常运动的事件,它们被细分为由突然的自主运动诱发的阵发性运动诱发性运动障碍(PKD);发作性非运动性运动障碍(PNKD),其在休息时发生;长时间运动后出现的阵发性劳累诱发的运动障碍(PED);以及发生在睡眠中的阵发性睡眠性运动障碍(PHD)。PxD可以是散发性的、家族性的(常染色体显性遗传)或继发于其他疾病。最近的遗传学发现可能有助于我们阐明这些疾病的病理生理学。PKD与16号染色体的着丝粒周区相关,PNKD与2号染色体长臂(2q32-36基因座)上的肌原纤维形成调节因子1 (MR-1)基因突变相关,PED与葡萄糖转运蛋白基因GLUT1突变相关,该基因负责葡萄糖转运穿过血脑屏障。
PxD其特征为反复发作的不自主肌张力障碍、舞蹈病、手足徐动症或其组合。通常根据触发因素分为三个亚组:阵发性运动诱发性运动障碍(PKD)、阵发性非运动诱发性运动障碍(PNKD)和阵发性运动诱发性运动障碍(PED)。睡眠期间阵发性运动障碍,以前被认为是第四种类型的阵发性运动障碍,现在被认为是夜间额叶癫痫的一种形式。

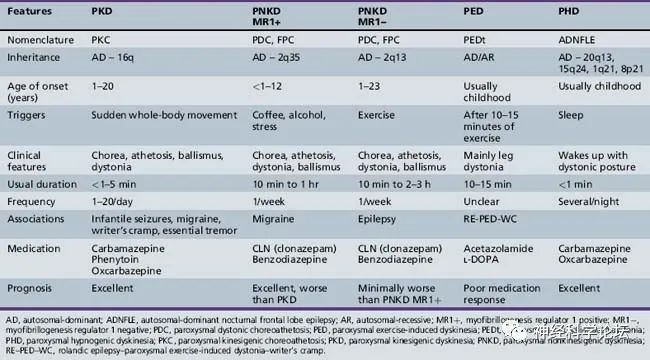
PKD是最常见的阵发性运动障碍类型,表现为在开始自主运动时突然出现短暂的反复发作。发作的持续时间通常比其他类型的短,通常持续不到1分钟。发病年龄通常在7至15岁之间,患者对卡马西平或奥卡西平等抗惊厥药表现出良好的反应。PNKD患者的发作通常持续时间较长(长达数小时),由酒精、咖啡或情绪压力引发。症状通常在20岁之前开始,治疗反应比PKD差。PED症状是由持续运动引发的,平均发病年龄为5岁,抗惊厥治疗似乎不是很有用。所有这三种类型的运动障碍通常与其他神经系统疾病有关,如婴儿癫痫发作、其他类型的癫痫、偏头痛、书写痉挛、共济失调或震颤。
自从2004年肌原纤维形成调节因子1基因(MR-1,OMIM 609023)首次被鉴定为PNKD的病因以来,已经发现了PKD的13个富含蛋白质的跨膜蛋白2基因(PRRT2,OMIM 614386)和PED的溶质载体家族2成员1基因(SLC2A1,OMIM 138140)。研究发现发作性运动障碍的表型和基因型重叠,以及亚型内的各种表现和表型异质性。发作性运动障碍可以根据触发因素分为三组,这种分类有助于诊断。然而,据报道,三组间的临床和遗传异质性(即使在同一表型内)以及表型-基因型重叠使得在某些情况下难以建立病原学诊断。此外,与PRRT2、SLC2A1和MR-1的作用相关的PKD、PNKD和PED的病理生理学尚未完全阐明,尽管最近有关于突触病变、转运病变和通道病变的研究报道。
随着基因测序的最新进展,与阵发性运动障碍相关的基因变异数量急剧增加,现在很明显存在显著的基因型-表型重叠、外显率降低(或不完全)和表型变异性。此外,多种遗传疾病可以表现为阵发性运动障碍作为初始症状。在一大型的发作性运动障碍儿童患者队列中研究了临床和遗传特征。正如所料,PKD是最常见的类型,并显示男性占优势,平均发病年龄为10.1岁。肌张力障碍是这些患者最常见的症状,持续时间短于1分钟,64.7%的患者在单独使用奥卡西平或卡马西平时达到无症状状态。5名PKD患者(12.5%)有BFIS病史,其中4名携带PRRT2突变。在13个检测的PKD家族中的9个(69.2%)和25个检测的散发病例中的8个患者(32.0%)中发现了6个PRRT2突变。最常见的突变是c.649dupC,占PRRT2阳性PKD患者的50%。
大多数以前的研究人员报道了PKD患者的临床特征以及PRRT2的遗传分析,据报道突变出现在18.5%至65%的病例中。最近的一项研究发现,在145例阵发性运动障碍患者中,PRRT2、SLC2A1和MR-1突变分别占35%、10%和2%,表明临床和遗传异质性,以及表型-基因型重叠。目前还没有涉及大量儿童阵发性运动障碍患者的遗传学或临床研究。在一项队列研究中,只有一名PRRT2阴性和SLC2A1阴性的阵发性运动障碍患者有常染色体显性家族史,即PKD伴癫痫,因此任何MR-1突变的可能性较低。PRRT2、SLC2A1和MR-1突变分别在18例(32.7%)、2例(3.6%)和0例儿童阵发性运动障碍患者中发现。
在另一个韩国队列的散发病例中没有携带这种突变,然而我们在62.5%的PRRT2阳性PKD散发病例中发现了这种突变。这代表了世界范围内散发性和家族性PKD病例患者中的热点突变。与之前的研究一致,35%的PKD患者(约22%的PRRT2阳性PKD患者)除了开始自主运动外,还有其他额外的触发因素,包括紧张、情绪紧张、寒冷暴露和感染。PRRT2阳性组和PRRT2阴性组之间的治疗反应没有显著差异,这与以前的研究一致,可能是由于不同遗传缺陷之间共有的一些病理机制。
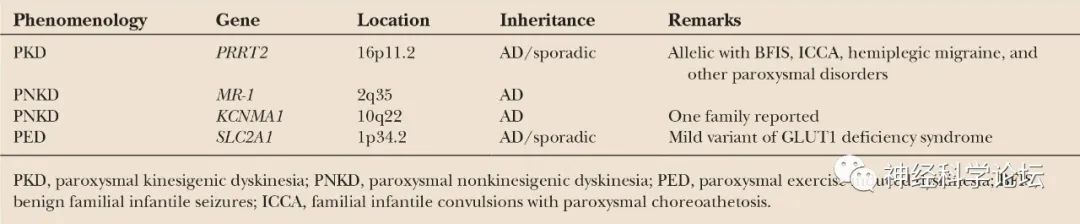
阵发性运动诱发性运动障碍(Paroxysmal kinesigenic dyskinesia,PKD):PKD是最常见的类型,并显示男性占优势。临床特征是由突然运动引起的短时间(几秒到几分钟)和频繁(每天多达100次)的张力障碍或舞蹈病发作。发病年龄通常是儿童期或青春期,但症状可以在成年后消失。PKD有一个已知的基因dyt 10(prr T2;富含脯氨酸的跨膜蛋白2)突变,PKD的第二个基因座被命名为DYT19。
阵发性非运动诱发性运动障碍(Paroxysmal non-kinesigenic dyskinesia,PNKD):以肌张力障碍、舞蹈病、舞蹈障碍或手足徐动症发作为特征。这些通常是由酒精或咖啡因引起的,但有时没有确定的诱因。发作持续几分钟到几小时,频率从每天一次到每年一到两次。发病年龄通常在儿童期或青春期,但这种运动障碍可能出现得足够晚,在50岁时发生。PNKD有一个已知的基因DYT8(PNKD1)和一个基因座DYT20 (PNKD2)突变。
PKD特征为反复和短暂的不自主运动发作,包括肌张力障碍、舞蹈病、舞蹈症或这些症状的组合,通常由突然的自主运动触发。长期以来,基底神经节-丘脑-皮质回路的紊乱被认为是不自主运动的原因。基底神经节的门控功能受损会导致向丘脑的异常输出,这又会导致大脑皮层的过度激活。已经在PKD患者中发现基底神经节、丘脑和皮质的结构和功能异常以及这些脑区之间的异常连接。最近的研究强调了小脑在PKD中的作用。从小脑皮质到小脑深部核的抑制不足会导致丘脑皮质通路的过度兴奋。
阵发性运动诱发的运动障碍(Paroxysmal exercise-induced dyskinesia ,PED):特征是运动诱发的肌张力障碍、舞蹈症和弹道运动发作,影响运动的肢体,持续几分钟到一小时。这种疾病通常在儿童期发病,并可有其他疾病表现,包括癫痫、偏头痛、发育迟缓和溶血性贫血。它显示了一种外显率略有下降的常染色体显性遗传模式。已知DYT18与编码葡萄糖转运蛋白1 (GLUT1)的SLC2A1基因突变有关。
PxD治疗基于非药理学和药理学方法的结合。对PNKD和PED有效的药物和非药物治疗也是可用的。对于传统治疗难以治愈的PxD,手术可能是一种替代治疗选择。PRRT2-PKD和MR-1-PNKD的病程是良性的,随着年龄的增长可能不需要治疗。改变生活方式以避免诱发因素在阵发性运动障碍的治疗中很重要。药物治疗尚未在对照试验中进行检验。然而,已发现抗惊厥药对治疗PKD病极其有效,有时对其他类型也有效,这表明这些疾病可能确实代表了通道病的形式。诸如乙酰唑胺、抗胆碱能药、左旋多巴和丁苯那嗪等药物一直不太成功。在罕见的医学难治性症状病例中,也采用了深部脑刺激。针对不同阵发性运动障碍的成功治疗的发展依赖于阐明病理生理学和靶向治疗以治疗潜在的扰动。
参考文献
Kim SY, Lee JS, Kim WJ, Kim H, Choi SA, Lim BC, Kim KJ, Chae JH. Paroxysmal Dyskinesia in Children: from Genes to the Clinic. J Clin Neurol. 2018 Oct;14(4):492-497. doi: 10.3988/jcn.2018.14.4.492.
Li ZY, Tian WT, Huang XJ, Cao L. The Pathogenesis of Paroxysmal Kinesigenic Dyskinesia: Current Concepts. Mov Disord. 2023 Apr;38(4):537-544. doi: 10.1002/mds.29326.
Sharma N. Neuropathology of Dystonia. Tremor Other Hyperkinet Mov (N Y). 2019 Feb 25;9:569. doi: 10.7916/d8-j6sx-b156.
Latorre A, Bhatia KP. Treatment of Paroxysmal Dyskinesia. Neurol Clin. 2020 May;38(2):433-447. doi: 10.1016/j.ncl.2020.01.007.
Ekmen A, Meneret A, Valabregue R, et al. Cerebellum Dysfunction in Patients With PRRT2-Related Paroxysmal Dyskinesia. Neurology. 2022 Mar 8;98(10):e1077-e1089. doi: 10.1212/WNL.0000000000200060.
Erro R, Magrinelli F, Bhatia KP. Paroxysmal movement disorders: Paroxysmal dyskinesia and episodic ataxia. Handb Clin Neurol. 2023;196:347-365. doi: 10.1016/B978-0-323-98817-9.00033-8.

