【绘真科普】多组学检测项目,如何辅助室管膜瘤患者分子分型?
时间:2024-06-04 16:00:22 热度:37.1℃ 作者:网络
室管膜肿瘤起源于脑室和脊髓中央管内衬的室管膜细胞(属于一种胶质细胞),约69%发生于儿童。由于室管膜瘤的组织学分型与其临床预后的相关性较差,2021年第5版WHO中枢神经系统肿瘤分类综合组织病理学、分子特征和解剖部位,共定义了10种不同ICD-O编码的室管膜瘤类型,用于辅助临床进行预后评估以及精准管理[1]。

图1 2021WHO中枢神经系统肿瘤分类室管膜瘤亚型及其ICD-O编码
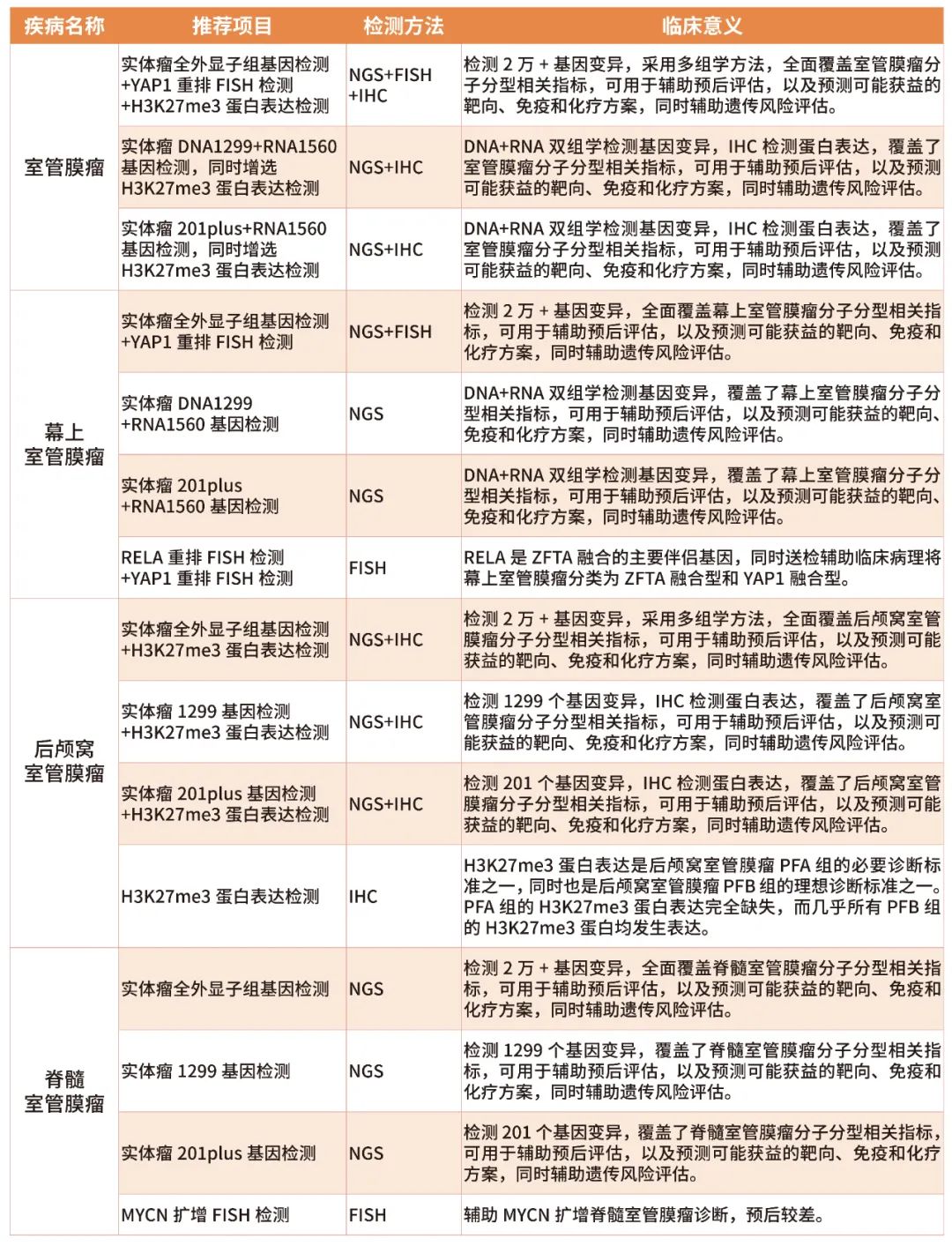
为此,绘真医学推出了系列检测项目,满足不同患者的诊疗需求。临床上,可根据患者的病灶位置以及组织病理学等信息,既可以送检多组学大中panel套餐,全面覆盖分子分型指标,用于预后评估,预测可能获益的靶向、免疫及化疗方案方案,同时辅助遗传风险评估,也可以送检单项小套餐,仅用于辅助分子分型及预后评估。若患者已经确诊为黏液乳头型室管膜瘤和室管膜下瘤,则建议按照实体瘤推荐基因检测项目。具体项目及其临床意义如表1所示。
表1 绘真医学室管膜瘤基因检测分子分型相关项目及其临床意义
室管膜瘤可发生于神经系统的任何部位,根据解剖学位置,分为后颅窝、幕上和脊髓。中山大学附属肿瘤医院研究团队纳入69例既往入院确诊的室管膜瘤患者,平均年龄为25岁,26例患者小于18岁。其中,28例(41%)患者病灶位于幕上区域,18例(26%)位于后颅窝,23例(33%)位于脊髓[2]。首都医科大学天坛医院研究团队入组了188例儿童和青少年室管膜瘤患者,中位年龄为5岁(0-17岁),65例(35%)患者病灶位于幕上,115例(61%)位于后颅窝,8例(4%)位于脊髓[3]。可见,随着年龄增加,脊髓室管膜瘤的占比显著增加。
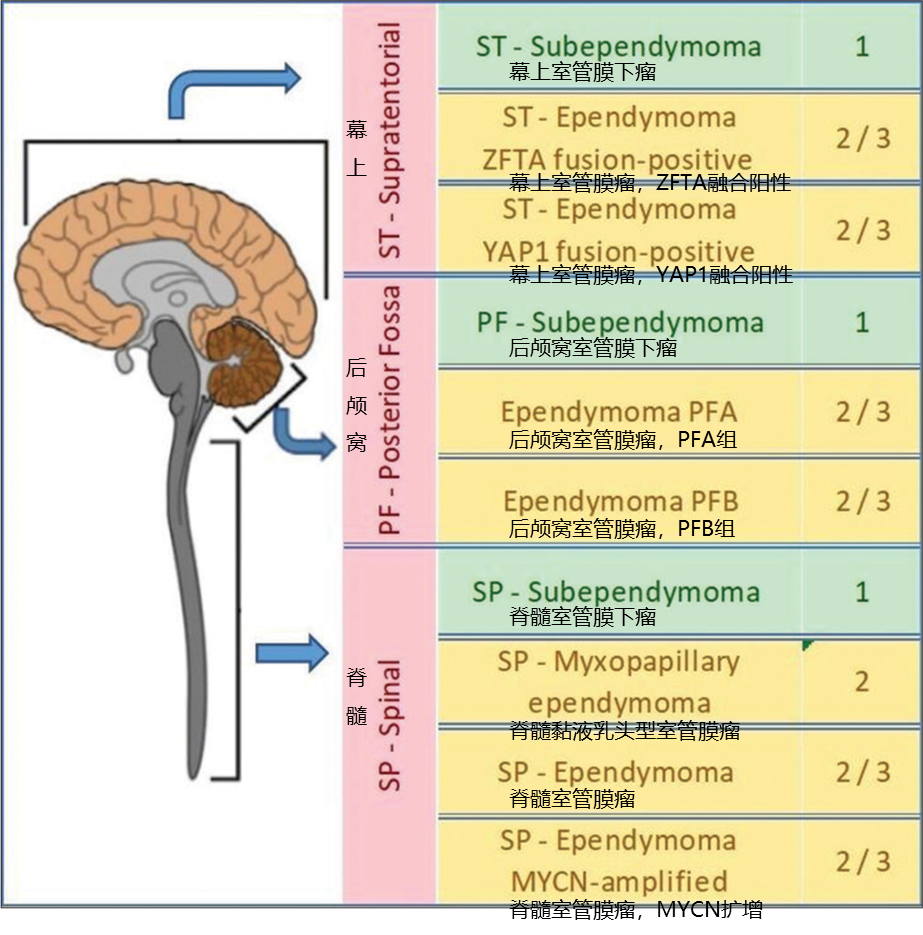
图2 室管膜瘤病灶位置及WHO分类分级
诊断所有室管膜瘤的先决条件是具有室管膜瘤的组织学和免疫组织化学特征。2021WHO分类定义的10种室管膜瘤亚型中,室管膜下瘤和黏液乳头型室管膜瘤均是基于组织形态学定义的类型。不同之处在于,室管膜下瘤可发生于幕上、后颅窝和脊髓,而黏液乳头型室管膜主要发生于脊髓,其余部位罕见。因此,在组织形态学排除室管膜下瘤和黏液乳头型室管膜后,其余亚型则在解剖部位的基础上进行分子分型诊断[4]。
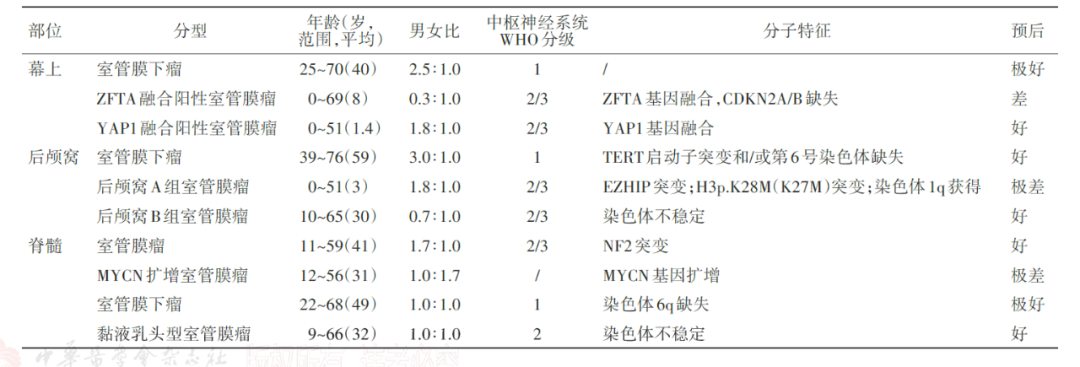
图3 第5版WHO分类室管膜瘤分类分级、临床病理分子特征及预后
一、幕上室管膜瘤
正如前文所述,已经确诊为室管膜瘤的患者,若组织形态学排除室管膜下瘤,同时病灶解剖部位为幕上,通常发生于额叶或顶叶,则归为幕上室管膜瘤。基于分子变异检测结果,又进一步细分为幕上室管膜瘤,ZFTA融合型和幕上室管膜瘤,YAP1融合型。若分子检测结果提示未携带ZFTA融合或YAP1融合,则归类为幕上室管膜瘤,NEC,这部分人群约占幕上室管膜瘤患者的17%-30%。若分子检测不能实现或失败,则归类为幕上室管膜瘤,NOS[5]。
ZFTA融合阳性幕上室管膜瘤,是室管膜瘤的主要亚型,在成人幕上室管膜瘤中的占比为20%-58%,在儿童中为66%-84%。RELA基因是主要融合伴侣,其他伴侣基因还有MAML2、NCOA1等。由于存在不同程度的间变性,ZFTA融合阳性幕上室管膜瘤被认定为CNS WHO 2-3级。临床研究表明,CDKN2A/B纯合缺失是ZFTA融合阳性幕上室管膜瘤患者的独立预后不良因素。
YAP1融合阳性幕上室管膜瘤并不常见,几乎只发生在儿童中。儿童队列数据显示,该亚型约占幕上室管膜瘤的6%-7.4%。MAMLD1基因是主要融合伴侣,其他伴侣基因还有FAM118B等。该亚型患者也表现出不同程度的间变性,故分级为CNS WHO 2-3级。现有临床研究表明,与ZFTA融合阳性相比,YAP1融合阳性幕上室管膜瘤患者预后相对较好。
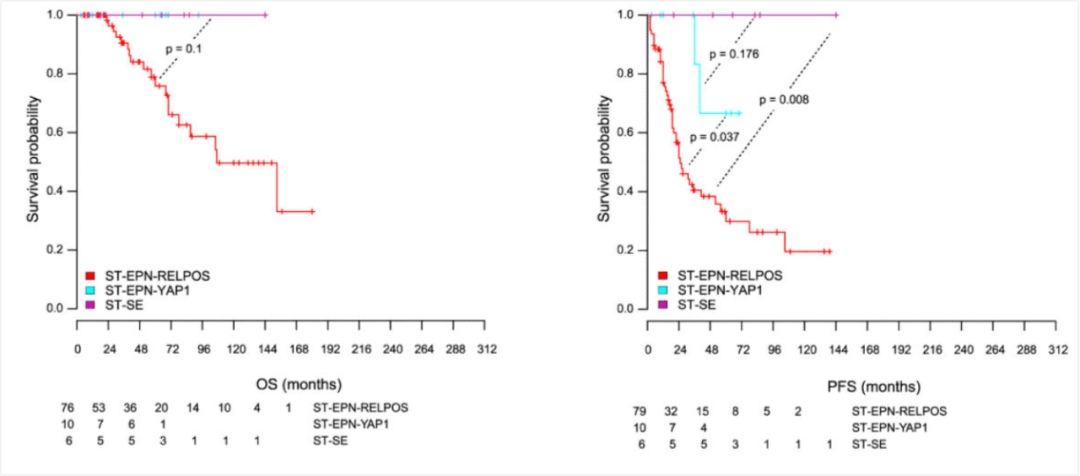
图4 发生于幕上的室管膜瘤患者生存状况比较,室管膜下瘤最佳,幕上室管膜瘤,YAP1融合阳性次之,幕上室管膜瘤,RELA融合阳性最差[6]
二、后颅窝(幕下)室管膜瘤
与幕上室管膜瘤定义相似,已经确诊为室管膜瘤的患者,若组织形态学排除室管膜下瘤,同时病灶解剖部位为后颅窝,则归为后颅窝室管膜瘤。肿瘤主要发生于第四脑室区,包括底部、外侧(小脑脚)和顶部,也可以发生于桥小脑脚。基于转录组或甲基化检测结果,后颅窝室管膜瘤又进一步细分为PFA组和PFB组。若分子检测结果并未确定具体亚型,则归类为后颅窝室管膜瘤,NEC。若分子检测不能实现或失败,则归类为后颅窝室管膜瘤,NOS[5]。
后颅窝A组室管膜瘤的诊断必要标准为后颅窝定位和肿瘤细胞核H3K27me3完全表达缺失,如果无法检测到H3K27me3完全表达缺失,与后颅窝A组室管膜瘤对应的DNA甲基化谱可以确定诊断。PFA组室管膜瘤主要发生在婴儿和儿童,中位年龄为3岁,95%确诊患者<6岁。由于表现出不同程度的间变性,后颅窝A组室管膜瘤被认定为CNS WHO 2-3级。
后颅窝B组室管膜瘤的诊断必要标准为后颅窝定位和与其一致的DNA甲基化谱。作为理想诊断标准之一,几乎所有的后颅窝B组室管膜均表达H3K27me3蛋白,只有非常罕见的患者表达缺失。PFB组室管膜瘤主要发生在成人,确诊时中位年龄为30岁。与PFA组相比,PFB组患者预后相对较好。
三、脊髓室管膜瘤
除组织形态学确诊为黏液乳头型室管膜瘤和室管膜下瘤外,其他定位于脊髓的室管膜瘤,根据是否检出MYCN基因扩增变异,分为脊髓室管膜瘤,NEC和MYCN扩增脊髓室管膜瘤。脊髓室管膜瘤沿椎管发生,好发部位依次为颈髓、胸髓和腰髓,常见于成人,中位确诊年龄为41岁[5]。
脊髓室管膜瘤,NEC,这类患者常携带NF2基因变异胚系或体系。若为NF2胚系有害变异,则提示该患者是由NF2型神经纤维瘤发展而来,具有遗传风险。大多数脊髓室管膜瘤被认定为CNS WHO 2级,预后相对较好。5-10年的PFS和OS分别为70%-90%和90%-100%。CNS WHO 3级患者罕见且预后差。
MYCN扩增脊髓室管膜瘤必要诊断标准为脊髓定位和MYCN扩增。这类患者罕见且预后差,迄今为止仅报道27例,预后差,75%-100%复发,中位生存期仅87个月。WHO尚未对该类患者进行明确分级。
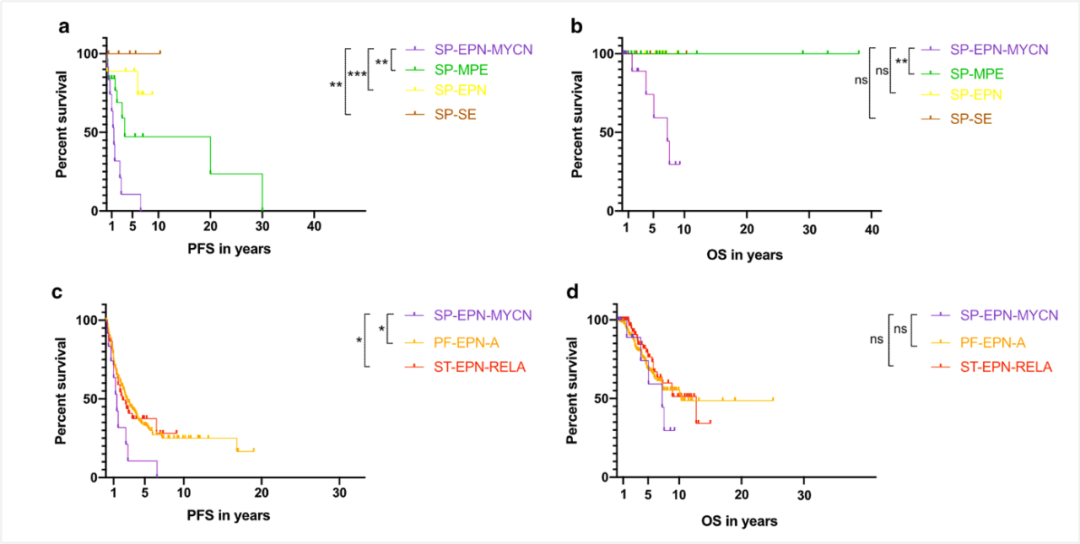
图5 发生于脊髓的室管膜瘤患者生存状况比较,室管膜下瘤最佳,其次是脊髓室管膜瘤,黏液乳头型室管膜瘤次之,脊髓室管膜瘤,MYCN扩增最差。另外,RELA融合阳性幕上室管膜瘤和PFA组后颅窝室管膜瘤预后也好于脊髓室管膜瘤,MYCN扩增型[7]
四、黏液乳头型室管膜瘤和室管膜下瘤
黏液乳头型室管膜瘤和室管膜下瘤,均是组织形态学定义的类型。前者主要发生在脊髓圆锥和终丝,其他部位罕见。后者可发生在中枢神经系统的所有脑室,常见于第四脑室和侧脑室。因此,对于幕上室管膜瘤和后颅窝室管膜瘤的分子分型,需要首先基于组织形态学特征,排除室管膜下瘤。脊髓室管膜需要先排除黏液乳头型室管膜瘤和室管膜下瘤[5]。
黏液乳头型室管膜瘤在儿童和成人中的预后均相对较好,10年总生存率超过90%。然而,约20%患者可原位复发或远处播散。鉴于此,新版分类将黏液乳头型室管膜瘤分级为CNS WHO 2级。室管膜下瘤总体预后极佳,即使次全切除也极少复发,WHO分级为1级。既往临床研究表明,后颅窝室管膜下瘤,若携带TERT启动子突变,PFS明显缩短,预后较差。
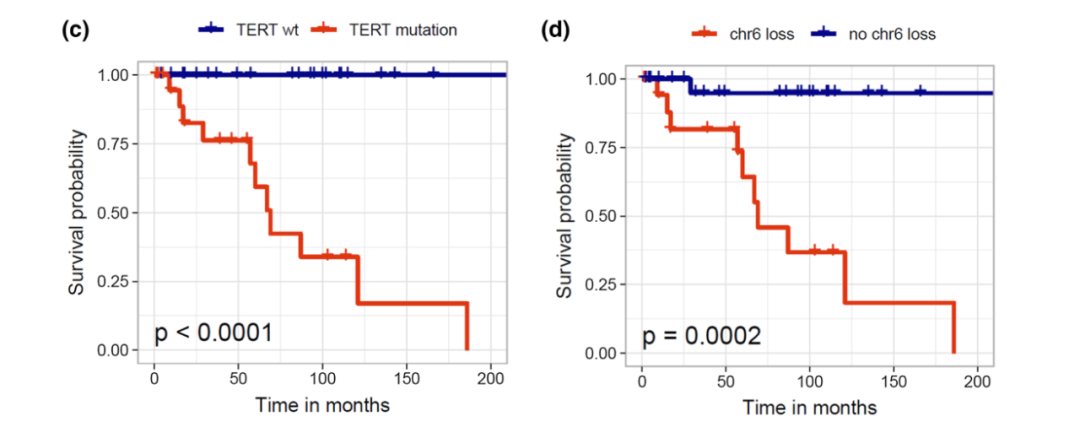
图6 临床研究发现,TERT启动子突变和6号染色体缺失变异,与后颅窝室管膜下瘤预后不良相关[8]
参考文献:
[1]申楠茜,张佳璇,甘桐嘉,et al.2021年WHO中枢神经系统肿瘤分类概述[J].放射学实践, 2021, 36(7):14.DOI:10.13609/j.cnki.1000-0313.2021.07.001.
[2]Xi, Shaoyan et al. “Clinical significance of the histological and molecular characteristics of ependymal tumors: a single institution case series from China.” BMC cancer vol. 19,1 717. 19 Jul. 2019, doi:10.1186/s12885-019-5877-9
[3]Han, Zhe et al. “Prognostic value of H3K27me3 in children with ependymoma.” Pediatric blood & cancer vol. 67,3 (2020): e28121. doi:10.1002/pbc.28121
[4]王行富,郑莉梅,张声. 第5版WHO中枢神经系统肿瘤分类室管膜肿瘤解读[J]. 中华病理学杂志,2023,52(03):223-227.DOI:10.3760/cma.j.cn112151-20220916-00781
[5]WHO Classification of Tumours Editorial Board. Central nervous system tumours.Lyon(France):International Agency for Research on Cancer;2021.(WHO classification of tumors series,5th ed.;vol.6).https://publications.iarc.fr/601
[6]Pajtler, Kristian W et al. “Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups.” Cancer cell vol. 27,5 (2015): 728-43. doi:10.1016/j.ccell.2015.04.002
[7]Ghasemi, David R et al. “MYCN amplification drives an aggressive form of spinal ependymoma.” Acta neuropathologica vol. 138,6 (2019): 1075-1089. doi:10.1007/s00401-019-02056-2
[8]Thomas, Christian et al. “TERT promoter mutation and chromosome 6 loss define a high-risk subtype of ependymoma evolving from posterior fossa subependymoma.” Acta neuropathologica vol. 141,6 (2021): 959-970. doi:10.1007/s00401-021-02300-8


