Nature子刊发表综述:CAR-T相关毒性的管理
时间:2024-08-14 14:01:31 热度:37.1℃ 作者:网络
CAR-T相关毒性的管理
CAR-T细胞疗法彻底改变了大B细胞淋巴瘤、多发性骨髓瘤、急性淋巴细胞白血病等多种血液恶性肿瘤的治疗,也正在多种实体瘤患者中进行研究。细胞因子释放综合征(CRS)和免疫效应细胞相关神经毒性综合征(ICANS)是CAR-T细胞相关特征性毒性,而随着支持治疗的改善以及免疫抑制剂的应用,使CAR-T细胞治疗更安全、更可行。但随着临床经验的增多,进一步发现一些既往不太明确的毒性,包括运动障碍、免疫效应细胞相关血液毒性(ICAHT)和免疫效应细胞相关噬血细胞性淋巴组织细胞增生症样综合征(IEC-HS),以及CAR-T细胞诱导的持续性B细胞再生障碍(B cell aplasia)和低丙种球蛋白血症患者发生感染的风险。目前正在使用更多样化的免疫抑制和支持治疗药物进行毒性管理,但尚无通用策略。此外,随着靶向新抗原的CAR-T细胞产品的开发,损伤表达靶抗原的非恶性组织的额外毒性也成为潜在障碍。毒性管理策略的持续前瞻性评估和低毒性CAR-T细胞产品的设计对于该领域的持续成功均至关重要。
《Nature Reviews Clinical Oncology》近日发表综述,阐述了对于CAR-T细胞疗法相关毒性的不断发展的认识及其临床管理。现整理全文供参考。

本文要点
• 随着临床医生在CAR-T细胞治疗方面获得更多经验,CRS的管理也得到改善,特别是使用IL-6受体拮抗剂托珠单抗。
• 此外,ICANS的监测和治疗也随着支持治疗和糖皮质激素使用的改进而得到改善。
• 运动障碍是B细胞成熟抗原(BCMA) CAR-T细胞的罕见并发症,其管理存在困难,可能危及生命。
• 长期血细胞减少、继发性噬血细胞性淋巴组织细胞增多症和感染性并发症越来越被人们所认识,并且已经制定了一致的治疗指南。
• CAR-T细胞治疗后报道新的T细胞恶性肿瘤,但非常罕见。CAR-T细胞治疗后应无限期监测患者是否发生第二恶性肿瘤。
• 该领域的未来发展方向包括开发毒性更低的CAR-T细胞产品。
CRS
CRS是一种炎症综合征,与CAR-T细胞和其他免疫细胞(如单核细胞和巨噬细胞)分泌炎性细胞因子(如IL-6、IFNγ和IL-2)有关,其典型体征和症状包括发热、寒战、窦性心动过速、低血压、缺氧和呼吸困难。CRS相关死亡可归因于呼吸衰竭、心脏骤停、多器官衰竭或继发性HLH,已见于多种不同CAR-T细胞产品(表1)。
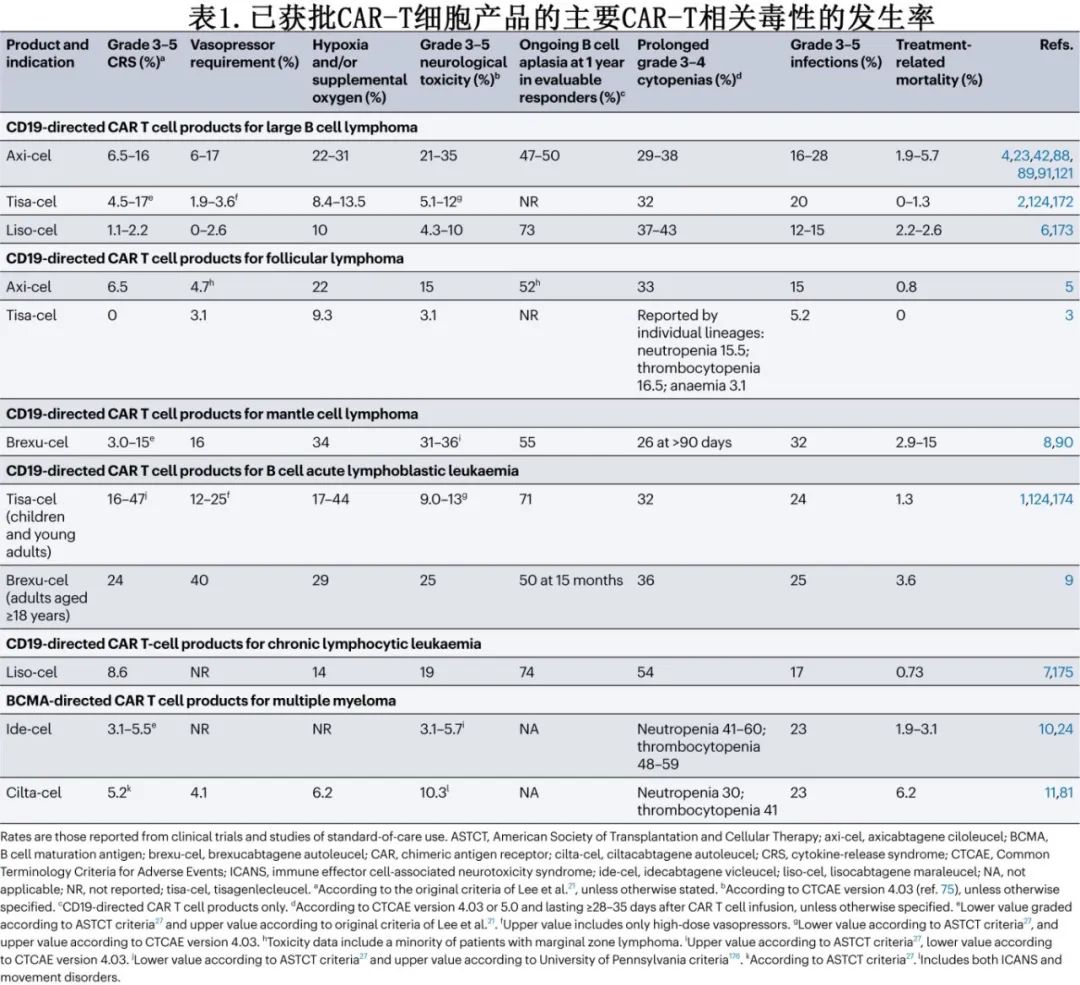
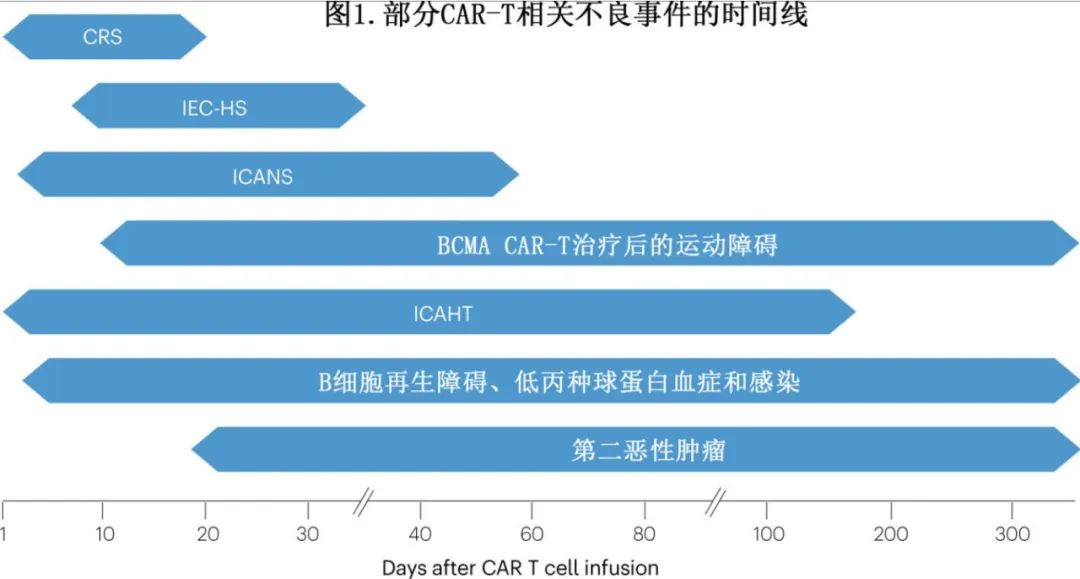
CRS多发生于CAR-T细胞输注后14天内(图1),中位至发作时间为2-7天,时间可能因多种因素而异,包括CAR-T细胞产品及其治疗的恶性肿瘤,但高级别CRS可在CAR-T细胞输注后数小时内开始。发热通常是CRS发病的标志,也是最常见的首发表现。在部分患者中,CRS可能难以鉴别或与其他毒性重叠:在与CRS相关高热期间观察到的轻度意识模糊可能反映或不反映早期ICANS,继发性HLH可能难以与重度CRS鉴别,尽管HLH可引起骨髓抑制,但在无HLH的情况下也可发生长期血细胞减少。
CRS的危险因素包括恶性肿瘤的高淋巴结和/或骨髓负荷、ECOG体能状态差、治疗前血清炎症标志物升高、基线低血小板计数、接受和/或需要桥接治疗、强化清淋化疗、高CAR-T细胞剂量和高CAR-T细胞峰值血液水平。重度CRS的风险取决于所使用的CAR-T细胞产品及其治疗的恶性肿瘤(表1)。目前已开发出多种风险分层模型来预测严重CRS的发生,它们通常包括清淋前实验室测试值和/或CAR-T细胞输注后血清中细胞因子或其他免疫蛋白的早期水平,如C反应蛋白(CRP)、铁蛋白、IL-6、颗粒酶B、fractalkine(亦称CX3CL1)、CC-motif趋化因子2 (CCL2,亦称MCP1)、CCL3 (MIP1α)、IFNγ、可溶性IL-1受体拮抗剂、嗜酸性粒细胞趋化蛋白(eotaxin)和可溶性糖蛋白130。鉴于内皮活化是CRS和ICANS的潜在机制,内皮活化和应激指数(EASIX)评分已用于重度CRS风险分层,其包括基线血肌酐、乳酸脱氢酶(LDH)和血小板水平,以及修改的EASIX公式。
在2019年美国移植和细胞治疗学会(ASTCT)共识指南发表之前已出现多个CRS分级系统,包括Lee等发表的原始标准。2014年,Penn-Pennsylvania大学分级系统用于tisa-cel的早期多中心试验,Memorial Sloan Kettering系统用于B细胞白血病患者CD19 CAR-T细胞的早期评价。但不同分级系统的使用导致研究和比较CAR-T细胞产品之间的CRS毒性变得复杂(表1)。然而随着最近的临床试验和CAR-T标准治疗试验中统一应用ASTCT量表,该异质性可能会在未来减少。ASTCT系统具有简单易用的优点,但该CRS分级系统仅反映体温、血压和呼吸状态的变化。因此,凝血功能障碍、心脏射血分数降低、肝或肾功能不全和电解质紊乱等器官毒性必须使用不良事件通用术语标准(CTCAE)单独报告。ASTCT 3级CRS是一个广义类别,包括短暂使用低剂量血管加压药的患者,以及需要高剂量单一血管加压药(加或不加血管加压素)数天的患者。
IL-6受体拮抗剂托珠单抗是CRS的主要一线治疗药物(表2)。随着时间的推移,启动托珠单抗的建议阈值也在发生演变,转而推荐该药物用于ASTCT 2级CRS患者和部分ASTCT 1级CRS患者(表3)。也可考虑托珠单抗用于治疗特定器官毒性,如急性肾脏或肝脏衰竭、心脏射血分数降低或严重电解质紊乱,但大多数发生重度器官毒性的患者已出现≥2级CRS。预防性使用托珠单抗可能增加神经毒性的风险,但托珠单抗治疗已确诊的CRS是否会增加ICANS的风险尚不清楚。与接受0-1剂托珠单抗的患者相比,接受≥2剂托珠单抗的患者发生任何级别和高级别ICANS的风险更高,但也更可能发生任何级别或高级别CRS,因此患者本身可能存在易患CRS和ICANS的基础因素。CRS小鼠模型已证实托珠单抗对CAR-T细胞的抗白血病活性无影响。对临床试验数据的回顾性分析表明,使用托珠单抗治疗已确诊的CRS对CAR-T细胞血液水平无影响。接受托珠单抗治疗CRS的患者的缓解率似乎与未接受托珠单抗的患者相当。此外,尽管无法获得大队列随机试验,但在回顾性分析中,托珠单抗似乎不会降低缓解持久性、无进展生存期(PFS)或总生存期(OS)。
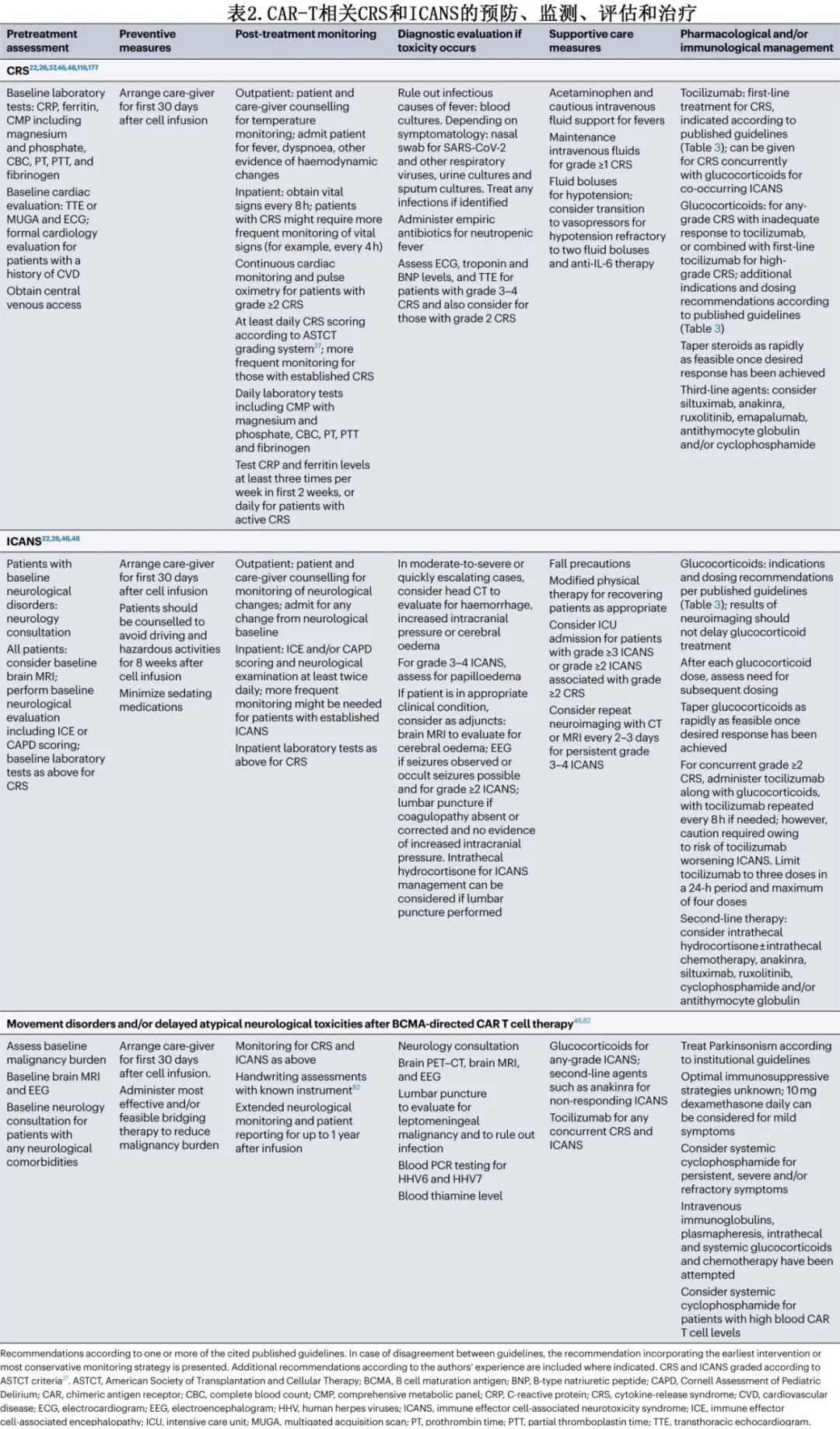
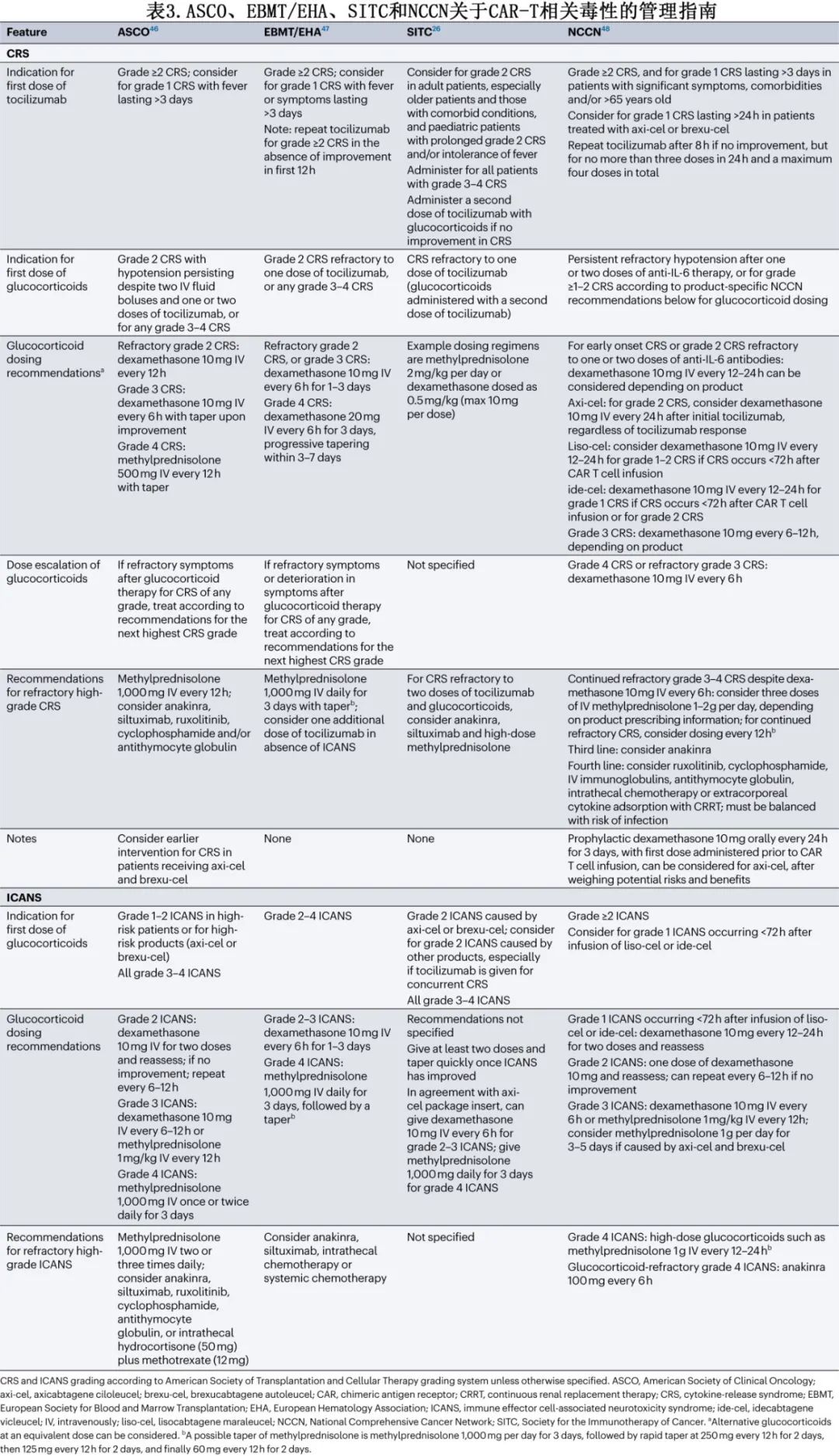
糖皮质激素是托珠单抗难治性CRS的标准治疗,通常为甲泼尼龙或地塞米松,需剂量递增,一旦达到毒性控制则立即停药或逐渐减量(如必要)(表3);但糖皮质激素的最佳剂量和给药时间尚不清楚。早期报告表明,患者接受大剂量糖皮质激素治疗炎症综合征(现称为CRS)后,血液CAR-T细胞水平降低、CAR-T细胞持久性差、恶性肿瘤复发。但其他回顾性分析未发现糖皮质激素对缓解率或持久性的有害影响。在一项对接受axi-cel治疗大B细胞淋巴瘤(LBCL)的患者进行的回顾性分析中,较高的累积糖皮质激素剂量与较短的PFS相关;此外,较早使用、较长期使用和较高的糖皮质激素累积剂量与较短的OS相关。当分析仅限于血清LDH水平升高(高肿瘤负荷替代指标)的患者时,该相关性仍得以维持。相比之下,纳入62例接受各种BCMA CAR-T细胞产品治疗的多发性骨髓瘤患者的回顾性分析中,任何使用或更高剂量的糖皮质激素对缓解率或生存结局均无影响,但糖皮质激素治疗≥5天与至下次治疗时间缩短相关。由于需要糖皮质激素治疗的患者可能存在其他生物学差异,使其容易发生重度毒性和对CAR-T细胞治疗反应较差,因此这些分析可能被混淆。尽管如此,糖皮质激素治疗的剂量和持续时间仍可能是确定其对CAR-T细胞活性和临床结局影响的关键因素。
很多专业协会发布了CRS管理原则,涉及托珠单抗、糖皮质激素和其他治疗,例如ASCO、ISCT、EBMT/EHA、SITC和NCCN(表3)。此外,FDA批准的CAR-T细胞疗法的说明书亦可为毒性管理提供产品建议。一般而言,上述原则中托珠单抗和糖皮质激素治疗的阈值相似。SITC指南首先发表(2020年),最普遍,NCCN指南则是最新的(2023年12月发布),较为具体,并纳入了最具体的产品特异性管理建议。
随着CAR-T细胞相关毒性的早期干预,高级别不良事件的发生率有所降低。例如在101例LBCL患者的II期ZUMA-1研究中,≥3级CRS和神经毒性的发生率均降低,可能部分归因于方案修订,允许更早给予托珠单抗或糖皮质激素。英国726例LBCL患者的最新数据表明,托珠单抗和糖皮质激素的使用频率高于2019年(CAR-T细胞项目的第一年),高级别CRS的频率较低(2020–2022年4%,2019年为9%;P=0.01),也可能是由于更有效的桥接治疗导致治疗前恶性肿瘤负荷降低。高级别CRS的减少与1年PFS和OS的改善相关(2020年-2022年的50% vs 2019 年的32%,60% vs 40%;P<0.001),表明毒性缓解策略的演变对CAR-T细胞治疗的疗效无影响。
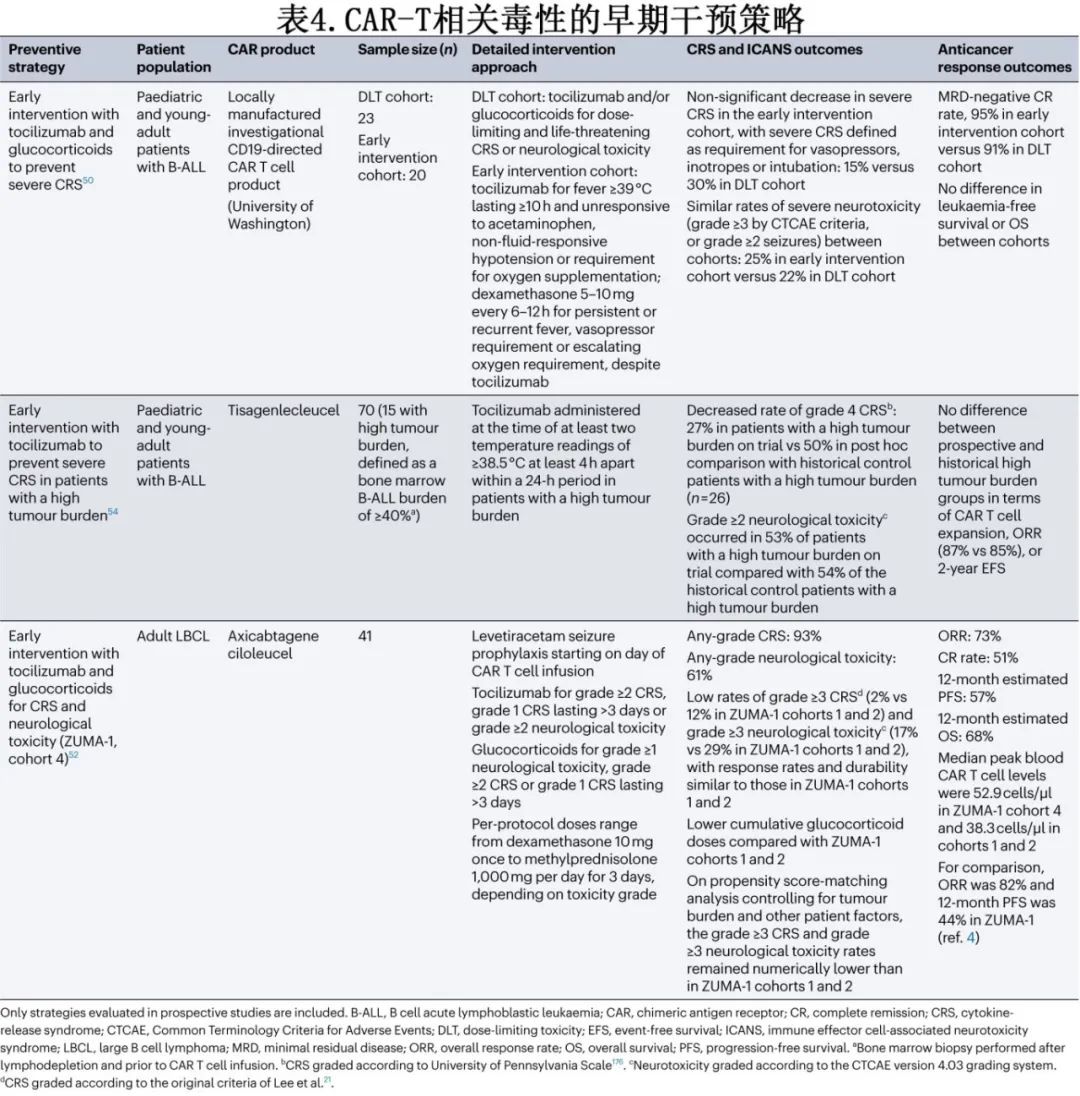
CD19 CAR-T细胞的前瞻性研究评价了CRS的多种早期干预(表4)和预防策略。托珠单抗和糖皮质激素预防性和(或)早期干预对降低高级别CRS的发生率有一定疗效,而对缓解率、缓解持久性或血液CAR-T细胞水平无明显不利影响。值得注意的是,对ZUMA-1队列3(在31例LBCL患者输注axi-cel后第2天预防性使用托珠单抗)数据的初步分析引起了对预防性使用托珠单抗是否会增加重度神经毒性风险的关注(41%的患者出现3-5级神经毒性,而ZUMA-1队列1和队列2为28%)。但在一项纳入20例LBCL患者的研究中,在第0天输注CAR-T细胞(表达4-1BB共刺激结构域)前1h预防性给予托珠单抗未观察到该趋势,仅1例患者出现重度ICANS。在其他分析中,使用托珠单抗作为单药治疗对CRS进行早期干预,或使用该药物联合糖皮质激素作为预防性和/或早期干预毒性管理策略,均未明显增加或降低重度神经毒性的风险(表4)。还在研究BTK抑制剂和JAK抑制剂用于CRS预防。
托珠单抗、小分子抑制剂和T细胞消融药物(ablative agents)以外的细胞因子拮抗剂作为类固醇难治性CRS的治疗药物,或作为托珠单抗难治性CRS患者的类固醇替代药物(steroid-sparing agents),其作用仍不明确。在缺乏随机试验的情况下,评估类固醇难治性CRS的新型治疗因以下情况而极为复杂:该毒性几乎总是随时间推移自然消退(或在使用更大累积剂量的糖皮质激素时),通常在尝试挽救治疗时继续。抗IL-6抗体司妥昔单抗和IL-1受体拮抗剂anakinra已用于管理CRS。anakinra在小鼠模型中可减轻CRS,而不影响CAR-T细胞的抗肿瘤活性。病例系列报告了类固醇难治性CRS患者接受anakinra给药后CRS消退。JAK可介导细胞因子(如IL-6和IFNγ)受体的下游信号转导,病例报告表明,小分子JAK抑制剂芦可替尼可用于治疗类固醇难治性CRS,不会完全消除CAR-T细胞扩增;然而体外证据表明,芦可替尼可抑制CAR-T细胞增殖,需要仔细评估最佳时机和剂量。获批用于治疗原发性HLH的抗IFNγ抗体emapalumab最近报道用于治疗类固醇和多种其他抗细胞因子疗法难治的CRS,临床前证据表明IFNγ阻断不会抑制CAR-T细胞的抗肿瘤活性。在临床前模型中,达沙替尼可通过抑制淋巴细胞特异性蛋白酪氨酸激酶Lck,可逆性地阻止CAR-T细胞增殖、细胞因子产生和溶细胞活性,但达沙替尼治疗类固醇难治性CRS的临床证据有限。T细胞消融疗法,如细胞毒化疗环磷酰胺、抗胸腺细胞球蛋白(一种来源于兔或马的抗体制剂,用人T细胞前体进行高免)和靶向CD52(在各种免疫细胞上高表达,包括成熟T细胞)的单克隆抗体,已纳入难治性CRS的治疗中,其中CAR-T细胞完全耗竭是可接受的结局;然而尚缺乏这些疗法的大规模研究。
神经毒性
ICANS
神经毒性仍是CAR-T细胞治疗的主要问题和障碍。ICANS的各种可能的神经系统症状包括脑病、呼吸困难和警觉性水平不同程度降低(从嗜睡到迟钝);较少见的症状包括伴全身或局灶性肌无力、肌阵挛和癫痫发作。2016年,因为发生脑水肿导致数例死亡,在B细胞急性淋巴细胞白血病(B-ALL)患者中测试CD19 CAR-T细胞产品JCAR015的II期ROCKET试验被停止,此外在接受获批CD19或BCMA CAR-T细胞产品的患者中也发生了致死性神经毒性。事实上,ICANS通常不仅发生在CD19 CAR-T细胞中,也可发生在BCMA CAR-T细胞中。ICANS通常与CRS同时发生或在CRS后不久发生(图1),且在CRS之前不太常见。也有报告CAR-T细胞输注后>3周开始的迟发性ICANS和无CRS时发生的ICANS。
CAR-T细胞具有极好的穿透血脑屏障(BBB)和进入脑脊液(CSF)的能力,但并非所有可检测到CSF CAR-T细胞的患者均发生临床显著神经毒性。此外,在重度ICANS患者中,BBB受到破坏,对全身细胞因子具有高渗透性。在ICANS患者和CAR-T细胞诱导神经毒性的非人灵长类动物模型中,部分促炎细胞因子(包括IL-6和IL-1)在CSF中的浓度可能高于血清中的浓度,表明CNS中局部产生这些炎性介质。发生≥3级神经系统事件的患者CSF中炎性细胞因子和免疫蛋白(如颗粒酶和CRP)水平高于发生0-2级事件的患者,提示这些蛋白参与发病过程。ICANS的危险因素与CRS的危险因素高度重叠,包括淋巴结和/或骨髓高肿瘤负荷、基线时炎症标志物血清水平升高、基线血小板计数低、接受桥接治疗或需要桥接治疗、CAR-T细胞剂量高(产品内比较)、血液CAR-T细胞的峰值和曲线下面积升高。此外,CAR-T细胞输注前有克隆性造血证据的患者发生重度ICANS的风险增加;这种相关性尚不十分清楚,但可能反映潜在的促炎状态。高级别神经毒性更常发生于高级别CRS患者中。与CRS一样,改良EASIX评分或EASIX评分联合基线血清CRP和铁蛋白值可用于预测重度ICANS的风险。
神经事件的发生率因CAR-T细胞产品而异(表1)。在进行倾向性评分匹配的法国DESCAR-T登记组回顾性分析中,接受axi-cel治疗LBCL的患者的1-2级和≥3级ICANS发生率高于tisa-cel(分别为34.9% vs. 19.1%和13.9% vs. 2.9%;P<0.001)。axi-cel和brexu-cel引起的神经毒性增加反映出CAR使用CD28衍生的铰链、跨膜和共刺激结构域,导致体内CAR-T细胞扩增特别迅速,并产生更高水平的细胞因子和其他免疫刺激蛋白。然而,CAR的特定结构特征影响CAR-T细胞功能的方式因整体结构而异。例如,具有CD28共刺激结构域以及CD8a铰链和跨膜区域的CAR与低水平的神经毒性相关。
已上市CAR-T产品的首次试验中,多数使用CTCAE 4.03版对神经毒性进行分级,但cilta-cel除外,其在CARTITUDE-1试验的II期部分使用 ASTCT ICANS量表对神经毒性进行分级。ASTCT ICANS分级量表包括认知评分,即免疫效应细胞相关脑病(ICE)评分,包括定向、命名、遵循指令、书写和注意力领域;<12岁的儿童可以使用Cornell儿童谵妄评估工具而非ICE评分进行评估。该共识ICANS分级量表还包括患者的警觉性水平以及是否存在运动缺陷、癫痫发作、颅内压升高和脑水肿,如果存在则为≥3级。
糖皮质激素仍是ICANS最常用的一线治疗药物,但尚不清楚开始糖皮质激素治疗的最佳时机。由于担心CAR-T细胞增殖和/或持续性可能降低,因此早期治疗方案规定,仅在神经毒性达到CTCAE≥3级时使用糖皮质激素。然而与CRS一样,地塞米松目前可用于2级ICANS和部分1级ICANS患者。预防性使用低剂量糖皮质激素和早期使用进行干预治疗神经毒性可能会降低重度神经系统事件的发生率,同时降低毒性管理所需的糖皮质激素累积剂量(表4)。来自病例系列的证据表明,鉴于腰椎穿刺可以安全进行,鞘内糖皮质激素可能在ICANS的管理中发挥作用,即使在系统性糖皮质激素难治性病例中也是如此。还在临床试验(例如NCT04975555)中研究使用司妥昔单抗作为ICANS的一线治疗。
CRS与ICANS同时发生的情况下需要谨慎管理,需参考ASCO和NCCN共识建议中的具体指南。由于托珠单抗可能存在恶化神经毒性的风险,糖皮质激素治疗ICANS可能优先于管理低级别CRS。对于CRS与1级ICANS重叠,托珠单抗可单独治疗,或与糖皮质激素联合给药用于治疗CRS伴≥2级ICANS,但应监测患者的神经毒性恶化,尤其是在重复给药后(表2)。糖皮质激素通常根据指南中推荐的ICANS分级给药,但在并发重度CRS和轻-中度ICANS的患者中,给予糖皮质激素的剂量应足以管理更严重的毒性。同时发生≥2级CRS和≥2级ICANS是重症监护监测的指征。
Anakinra在小鼠模型中表现出预防CAR-T细胞诱导的神经毒性的活性,前瞻性临床研究的数据也表明可有效预防ICANS。在CAR-T细胞疗法的初始关键试验中,仅少数患者使用Anakinra治疗活动性ICANS,但其目前通常用作糖皮质激素难治性ICANS的标准治疗。尽管如此,Anakinra是否可以作为类固醇替代药物仍不清楚,病例系列数据表明,其使用未加快类固醇减量。与CRS相似,T细胞耗竭治疗(如细胞毒化疗和T细胞靶向药物)可能在糖皮质激素难治性、Anakinra难治性ICANS患者中发挥作用,以完全根除CAR-T细胞。
BCMA CAR-T细胞治疗后的运动障碍
在评价cilta-cel作为多发性骨髓瘤四线治疗的Ib/II期CARTITUDE-1研究中,12%的患者发生非典型迟发性神经综合征,中位至发作时间为27天(范围11-914天),而ICANS为5-9天。6%发生运动障碍,其表现包括帕金森症、书写困难、震颤、步态障碍、齿轮样强直、精神运动发育迟滞和情感贫乏(即情感表达严重受损或无表达);1例患者发生致死性帕金森症。其他迟发性神经系统不良事件包括精神状态改变、注意力损害、面瘫、复视、眼球震颤、颅神经麻痹、共济失调、感觉缺失以及周围运动和感觉神经病变,其发作为隐匿性,以小书写症为早期表现,且发生于ICANS消退后。这些非典型神经毒性通常对卡比多巴和/或左旋多巴、糖皮质激素或其他免疫抑制治疗无反应。在ide-cel治疗后也报告了帕金森症,表明运动障碍可能是BCMA CAR-T细胞产品的类效应。
在接受cilta-cel治疗的患者中确定的运动障碍危险因素包括以下两种或多种:基线高肿瘤负荷(定义为骨髓浸润伴恶性浆细胞≥80%,血清 M 蛋白≥5 g/L或受累血清游离轻链≥5,000 mg/L)、任何等级ICANS、≥2级 CRS 或血液 CAR-T 细胞高峰值和持续水平(第56天分别为>1000个细胞/µl和>300个细胞/µl)。在接受 cilta-cel 治疗后出现运动障碍的2例患者的尸检显示,二者均出现局灶性胶质细胞增生和基底神经节 T 细胞浸润,但没有黑质色素脱失(常见于原发性帕金森病患者);此外,在非恶性脑组织中未检测到 BCMA 表达。然而,关于相关脑区中 BCMA 表达的结果相互矛盾。在接受 cilta-cel 后出现帕金森症状的患者中,在基底神经节的尾状核神经元和星形胶质细胞亚群以及邻近额叶皮质的神经元中检测到 BCMA 表达(通过免疫组织化学评估),在 Allen Brain Atlas 的尾状核标本中也检测到 BCMA RNA 表达。相比之下,随后对来自63例无多发性骨髓瘤的供者的大脑、基底神经节、小脑和脑干样本进行的免疫组化检查未检测到 BCMA 表达(非特异性抗体结合除外)。因此,这些神经毒性是否为“on-target”毒性尚不明确。
在 cilta-cel 临床开发项目中实施加强监测和预防措施后,这些迟发性神经毒性的发生率降至<1%。管理策略包括更积极的桥接治疗以降低清淋前疾病负荷、输注后监测和笔迹评估、改良的早期干预策略以缓解 CRS 和ICANS(包括糖皮质激素用于任何级别 ICANS,如果并发任何级别 CRS 同时则联合托珠单抗)以及将神经毒性监测延长至细胞输注后1年(表2)。此外有证据表明,大剂量全身性环磷酰胺可减少血液中 BCMA CAR-T 细胞的极高数量(在细胞输注后>2周检测到),从而有效逆转迟发性神经毒性;然而其毒性仍是使用的潜在障碍。
血液学毒性
CAR-T相关骨髓抑制
清淋化疗后几天至一个月内发生的高级别血细胞减少非常常见,且符合预期。中性粒细胞减少与 CRS 重叠通常可导致中性粒细胞减少性发热,在接受 CAR-T 细胞治疗的患者中发生率为2–36%;但持续时间较长的≥3级血细胞减少通常发生于以B 细胞靶向和浆细胞靶向 CAR-T 细胞产品中(表1)。此外,在一小部分患者中报告了持续≥6个月的长期3-4级血细胞减少(主要是中性粒细胞减少和血小板减少),CD19 CAR-T 细胞输注后持续>6个月的中性粒细胞减少和血小板减少报告率分别约为1-4%和0-6%。在霍奇金淋巴瘤患者接受 CD30 CAR-T 细胞治疗后,甚至在实体瘤 CAR-T 细胞治疗后,也报告了长期血细胞减少,表明骨髓抑制不限于特定靶抗原。此外,血细胞减少,尤其是中性粒细胞减少,可以是双相或间歇性,在明显消退后数周至数月再次复发。
CAR-T 细胞治疗后长期骨髓抑制的病理生理学尚不完全清楚,风险因 CAR-T 细胞产品和预处理化疗方案而异,且该风险还随基线恶性肿瘤骨髓负荷升高、基线血细胞计数降低、基线血清炎症标志物水平升高、既往治疗次数增加、血液 IL-6 峰值水平升高以及 CRS 和/或 ICANS 加重而增加。这些相关性表明,造血储备、基线炎症环境和 CAR-T 细胞诱导的炎症均有助于长期血细胞减少。在前瞻性骨髓活检采样的病例系列中,治疗后1个月骨髓中高水平的炎性细胞因子和 CAR-T 细胞与长期骨髓抑制相关。骨髓样本的单细胞 RNA 测序显示,长期血细胞减少患者持续存在克隆扩增的表达IFNγ的CX3CR1highCD8+ T细胞群,并且骨髓中的长期IFNγ暴露可能抑制造血干细胞自我更新和分化。
CAR-T 治疗后发生的血细胞减少目前被 EHA 和 EBMT 称为免疫效应细胞相关血液毒性 (ICAHT)。晚期 ICAHT 定义为细胞输注后中性粒细胞持续减少超过30天,并根据中性粒细胞减少的严重程度和持续时间提出了早期和晚期 ICAHT 的分级量表。
ICAHT 的管理在不断发展中(表5)。

CAR-HEMATOTOX评分是一个5项工具,包括基线血红蛋白、血小板和中性粒细胞水平(与造血储备相关的标志物)以及炎症标志物铁蛋白和 CRP基线血清水平,可预测接受 CAR-T 细胞治疗的多发性骨髓瘤或各种淋巴瘤亚型患者的长期血液毒性和重度感染;因此,临床医生可考虑在 CAR-HEMATOTOX 评分较高的患者中进行基线骨髓活检、早期生长因子支持(从细胞输注后第2天左右开始),以及在中性粒细胞减少时开始对革兰氏阴性细菌和真菌感染进行抗菌预防。在 CAR-HEMATOTOX 评分较低的患者中,生长因子支持可延迟给药,直至中性粒细胞绝对计数降至<500个细胞/µl,并且可以避免预防性使用抗菌药物,但尚未对该策略进行前瞻性评价。≥3级 ICAHT 患者应接受阶梯评价,以排除骨髓抑制的其他原因(表5),可包括骨髓活检及随后的细胞遗传学和二代测序分析,并评价潜在的病毒病因,如巨细胞病毒 (CMV)、EB病毒或细小病毒感染。骨髓样本评估可排除其他原因,如 B 细胞或浆细胞恶心肿瘤进展,或发生新的髓系恶性肿瘤。晚期 ICAHT 患者的骨髓评估通常显示非特异性再生障碍和血细胞减少,应根据患者状态和机构指南输注红细胞和血小板。关于 CAR-T 细胞治疗后生长因子支持(即重组形式的粒细胞集落刺激因子)是否使 CRS 和/或 ICANS 恶化,回顾性报告存在矛盾;然而,即使有恶化风险,该风险也必须与长期中性粒细胞减少相关的感染风险进行平衡。
机制原理支持血小板生成素受体激动剂 (TPO-RA) 在治疗 CAR-T 相关血血细胞减少中的潜在效用,因为这些小分子激动剂可以绕过IFNγ对血小板生成素受体的抑制。尽管关于 TPO-RA 在这种情况下的数据有限,但仍越来越多地用于 CAR-T 细胞治疗后长期血细胞减少的患者。例如,来自美国的真实世界数据表明,15%的患者在接受 ide-cel 标准治疗后接受 TPO-RA 治疗血细胞减少。研究还尝试使用糖皮质激素管理 CAR-T 细胞血液毒性,但疗效尚不明确。在可获得冷冻保存细胞的患者中,自体干细胞加强(boost)疗法治疗长期血细胞减少已证实在 CD19 和 BCMA CAR-T 细胞治疗后的可行性;该策略也越来越多地使用,美国5%接受标准疗法治疗的患者还接受干细胞加强疗法治疗血细胞减少。异基因造血干细胞移植 (allo-HSCT) 是管理 CAR-T 细胞血液毒性的最后手段。
继发性HLH或IEC-HS
在接受 tisa-cel 治疗B-ALL、axi-cel治疗 LBCL 以及 ide-cel 或 cilta-cel 治疗多发性骨髓瘤的患者中报告了继发性HLH;在使用axi-cel、ide-cel和 cilta-cel 时还发生了罕见的致死性病例(发生率≤1%)。CAR-T相关HLH 是一种危及生命的巨噬细胞活化高炎症综合征,表现为血清铁蛋白水平高度升高、高甘油三酯血症、血细胞减少、凝血障碍、肺功能受损和肾和/或肝功能障碍,在部分患者中似乎是CRS 谱的重度终点。CARTOX小组最初提出 CAR-T相关继发性 HLH 的诊断标准、分级量表和管理指南,包括递增给予托珠单抗、递增剂量糖皮质激素和依托泊苷。CD22 CAR-T细胞的临床试验发现CRS改善或消退后发生的继发性 HLH 的独特模式,似乎具有不同的病理生理学。但CAR-T细胞相关 HLH 很少发生在既往无CRS的情况下,因此这两种综合征可能具有共同的机制。此外,HLH的确诊通常不需要骨髓活检评估噬血细胞;因此在许多患者中,严格区分重度 CRS 和 HLH 可能很困难。CAR-T细胞输注时既存NK细胞淋巴细胞减少与随后发生的 HLH 相关,可能是由于未抑制CD8+ T细胞过度活化,导致CD8+ T细胞无限制扩增和 CAR-T 细胞群收缩延迟。
在ASTCT支持下,CAR-T细胞相关继发性 HLH 的定义、分级系统和管理的共识建议已发表,提出术语“免疫效应细胞相关噬血细胞性淋巴组织细胞增生症样综合征”(IEC-HS)来描述该疾病。IEC-HS的病理生理学与 CRS 具有许多相同的特征,包括重叠的细胞因子谱,但根本诱因仍在研究中。IEC-HS的共识定义要求血清铁蛋白水平升高至正常上限的两倍以上或迅速升高,但也包括其他典型结果,如血细胞减少、转氨酶升高、低纤维蛋白原血症或噬血细胞的组织学证据。需要注意的是,除噬血细胞作用外,所有这些结果也可发生于CRS。Anakinra联合或不联合糖皮质激素均可有效治疗部分患者的 CAR-T 细胞相关HLH,并且 ASTCT 共识指南推荐其作为 IEC-HS 的一线治疗(表5)。事实上,Anakinra和糖皮质激素通常以递增剂量用于一线和二线治疗,芦可替尼、低剂量全身性依托泊苷和emapalumab是替代药物,但 IEC-HS 的管理仍在不断发展。
免疫毒性和感染风险
B 细胞再生障碍和低丙种球蛋白血症
B细胞再生障碍是接受CD19 CAR-T细胞治疗的患者免疫功能低下的重要原因;B细胞恢复率与持续性再生障碍不同,具体取决于 CAR-T 细胞产品和治疗的肿瘤(表1)。在无癌症复发的情况下,接受 axi-cel 治疗非霍奇金淋巴瘤 (NHL)(69-72%)、tisa-cel治疗 NHL(67%) 或接受 brexu-cel 治疗套细胞淋巴瘤 (62%) 或成人 B-ALL(83%) 的患者经常可获得 B 细胞恢复。然而,tisa-cel治疗后的 B 细胞恢复(特别是在细胞输注后6-12个月内)是儿童或年轻成人 B-ALL 患者疾病复发风险高的生物标志物。长期 B 细胞再生障碍是接受过 CAR-T 细胞治疗的患者疫苗应答的重要决定因素;例如,持续性 B 细胞再生障碍患者可能对 SARS-CoV-2 疫苗无体液应答,但可能获得T细胞免疫应答。
低丙种球蛋白血症常见于接受 CD19 和 BCMA CAR-T 细胞治疗的患者中,接受免疫球蛋白替代治疗的患者中比例则不同。CD19 CAR-T 细胞治疗后可发生 B 细胞和免疫球蛋白水平的自然恢复,但在数年内缓慢恢复。即使在B 细胞再生障碍持续的情况下,在 CD19 CAR-T 细胞治疗后仍可保留一定水平的体液免疫,这可能是由于长期存在的 CD19 阴性浆细胞所致,表现为抗病毒抗体的持续产生。然而,在 CD19 CAR-T 细胞治疗后第一年,对某些病原体的体液免疫可能降低。此外,LBCL患者在接受 CD19 CAR-T 细胞治疗后的前6个月或更长时间内对肺炎球菌结合疫苗缺乏体液应答,证明疫苗接种应答可能受损,但部分患者在第+360天接种疫苗后可产生体液保护作用。接受 BCMA CAR-T 细胞治疗的患者体液免疫可能更受损,因为长寿的 CD19 阴性、BCMA阳性浆细胞可能被耗尽,导致长期低丙种球蛋白血症的发生率更高。
尚缺乏支持免疫球蛋白替代治疗在预防 CAR-T 相关感染中作用的确切证据。回顾性数据表明,在接受BCMA×CD3双特异性抗体的多发性骨髓瘤患者中,静脉注射免疫球蛋白替代治疗可显著降低3-5级感染,但在 CAR-T 中该问题仍未得到充分研究。建议在 BCMA CAR-T 细胞治疗后、儿童在接受 CD19 CAR-T 细胞治疗后(由于浆细胞库不太稳健)、成人在CD19 CAR-T 细胞治疗后的前3个月以及发生复发性感染的任何 CAR-T 细胞受者中,进行免疫球蛋白替代治疗,目标为>400mg/dL。
感染
CAR-T 细胞治疗后常见≥3级细菌、病毒和真菌感染,估计有5-32%的患者发生(表1)。感染相关死亡是 CAR-T 细胞治疗后死亡的主要原因,中位随访持续10-16个月时为1-12%。CAR-T细胞治疗后感染的易感因素是多因素的,包括由清淋化疗诱导的CD4+ T细胞淋巴细胞减少(可持续>1年)、CD19 CAR-T 细胞引起的 B 细胞再生障碍、BCMA CAR-T 细胞引起的浆细胞耗竭、用于毒性管理的免疫抑制治疗以及与血液恶性肿瘤和既往癌症治疗相关的其他基础免疫缺陷。糖皮质激素和其他免疫抑制治疗可以抑制患者发热症状或其他感染体征,需要根据机构指南监测隐匿性感染(例如监测血培养)。CAR-T细胞治疗后感染的治疗前危险因素包括恶性肿瘤类型(B-ALL高于NHL)、既往治疗方案数量较多、既往allo-HSCT、接受桥接化疗和 CAR-HEMATOTOX 评分较高。
托珠单抗是否增加接受 CAR-T 细胞治疗患者的感染风险的报道相互矛盾,其可能并非独立的风险因素。然而,然而,中性粒细胞绝对计数<500细胞/μl持续≥14天和细胞输注后第0天至第21天使用地塞米松等效剂量10mg/天的糖皮质激素持续≥9天是CAR-T细胞治疗后第一个月发生高级别感染的独立危险因素,但该风险可以通过革兰氏阴性抗菌药物预防来改善。
CAR-T 细胞治疗后可发生多种病毒感染,其中呼吸道病毒是最常见的病因。在使用抗 CD52 抗体进行密集清淋后,CMV反应尤其值得关注,并且在异基因“现成”CAR-T细胞产品的试验中已有报告,其中使用CD52 抗体来预防异基因细胞的排斥反应。在接受 CD19 CAR-T 细胞产品的患者中,JC多瘤病毒 (JCPyV) 和人类疱疹病毒6型 (HHV-6) 再激活可导致死亡,原因在于在明显清淋的情况下出现病毒性脑病。CD19 CAR-T 细胞输注后1年发生致死性乙型肝炎病毒 (HBV) 再激活的报告表明,由于持续性 CAR-T 细胞介导的 B 细胞耗竭,HBV感染患者可能需要无限期继续预防性 HBV 抗病毒治疗。对于几乎所有临床使用的 CAR-T 细胞产品,均使用γ-轮状病毒或慢病毒对 T 细胞进行基因修饰;幸运的是,CAR-T细胞输注后在患者中未检测到具有复制能力的γ逆转录病毒或慢病毒。
随着 SARS-CoV-2 的出现,CD19 CAR-T和BCMA CAR-T后均发生了COVID-19感染相关死亡。既往接受过 CAR-T 细胞治疗的患者发生重度感染和死于 COVID-19 的可能性增加,CAR-T受者 COVID-19 死亡率的历史估计值为33–41%,但奥密克戎变种所致死亡风险的最新估计值显著较低,为7%。CAR-T受者也有较高的迁延性 COVID-19 和持续性病毒排出的风险,且对 SARS-CoV-2 疫苗的体液应答较差,尽管免疫可以随着加强针而增加;对疫苗的 T 细胞介导的免疫应答可提供部分针对重度 COVID-19 的保护。由于接受 CAR-T 细胞治疗的患者特别容易感染SARS-CoV-2,因此在细胞输注前后接种疫苗、使用现有最佳预防策略预防重度感染和对活动性感染患者采取合理的治疗策略至关重要(表5)。然而,预防和早期干预感染相关不良事件仍在发展,且各中心之间存在很大差异(表5)。
越来越重要的毒性
表达靶抗原的非恶性组织受损
由于CAR-T 中新抗原靶点的增加,以及需要为CAR-T治疗后复发的血液系统恶性肿瘤患者提供替代疗法,CAR-T损伤同样表达靶抗原的非恶性组织(即所谓的“on-target,off-tumour”效应)的可能性也在增加。GPRC5D CAR-T在多发性骨髓瘤患者中表现出良好的疗效,包括 BCMA CAR-T细胞治疗后复发的患者。GPRC5D主要由非恶性和恶性浆细胞表达,但在皮肤毛囊和硬角化上皮组织中也低水平表达。因此,GPRC5D CAR-T的预期毒性包括可控的皮疹、指(趾)甲变化、味觉障碍和口干,但也描述了一种重度小脑疾病,表现为头晕、共济失调、构音障碍和视觉固定困难,由CAR-T细胞靶向延髓橄榄核中表达GPRC5D的细胞所致;其似乎与剂量相关,在较低的细胞剂量下可避免。
血液学疾病中开发治疗 B 细胞和浆细胞恶性肿瘤以外的CAR-T 细胞疗法具有挑战性,因为其具有消除非恶性造血和免疫细胞的有害作用,可能需要通过 allo-HSCT 进行造血解救。设计治疗髓系恶性肿瘤的 CAR-T 细胞的抗原靶点均不理想,例如在非恶性造血干细胞和髓系祖细胞上表达的 CD33 和CD123;在 B 细胞、T细胞和自然杀伤细胞上表达的CD38;以及在肺和上皮细胞上表达的 C 型凝集素样分子1(CLL1,也称为 C 型凝集素结构域家族12成员a)。
由于缺乏表达仅限于恶性细胞的靶抗原,CAR -T细胞治疗实体肿瘤极具挑战性。例如,由于间皮素在肺上皮细胞上的表达,高剂量的间皮素靶向CAR-T细胞具有显著肺毒性。
第二原发恶性肿瘤的风险
髓系恶性肿瘤。已知CAR-T细胞治疗后可发生继发髓系恶性肿瘤,但通常归因于既往化疗方案和/或自体HSCT,而非CAR-T细胞产品本身。一项包含449例在宾夕法尼亚大学接受FDA批准的CAR-T细胞疗法的成年患者的队列回顾性分析显示,5年期间第二实体恶性肿瘤的估计风险为15.2%,第二血液恶性肿瘤的风险为2.3%,这与非CAR-T细胞疗法在此类患者的预期发生率相似。然而,在参与CARTITUDE-1试验的97例患者中有10例(10.3%)发生第二髓系肿瘤后,2023年12月FDA要求cilta-cel说明书中增加关于“继发性血液恶性肿瘤,包括骨髓增生异常综合征和急性髓系白血病”的黑框警告,表明监管机构和行业担心CAR-T相关继发恶性肿瘤风险可能增加。尽管难以进行直接比较,但CARTITUDE-1队列中第二髓系恶性肿瘤的发生率在数值上高于多发性骨髓瘤患者既往报告的5-7%。接受CD19 CAR-T细胞治疗LBCL的患者数据表明,清淋前克隆性造血易诱发第二髓系恶性肿瘤的后续发展(24个月累积发生率为19%,而无克隆性造血的患者为4.2%;P=0.028)。随着 CAR-T 细胞疗法在血液恶性肿瘤治疗方案中的应用越来越早线,由于患者既往化疗暴露减少,第二髓系恶性肿瘤的风险可能会降低。
T细胞恶性肿瘤。2023年11月,FDA发布新闻稿称,正在调查CAR-T细胞治疗后发生T细胞恶性肿瘤的风险,该风险适用于所有6种市售CAR-T细胞产品,并建议对接受CAR-T细胞治疗的患者进行终生监测。截至2023年12月31日,共向FDA报告了22例CAR-T细胞治疗后发生T细胞恶性肿瘤的患者,6种获批产品中有5种报告了病例。在其中3例T细胞恶性肿瘤的恶性克隆中检测到CAR转基因,但未获得所有患者的基因测序数据
根据已发表的 CAR-T 细胞源性恶性肿瘤病例报告,2例接受研究性异基因、供者源性 CD19 CAR-T 细胞(使用 PiggyBac 转座子系统进行基因修饰)的患者发生CAR+ T细胞淋巴瘤,但未发现转基因整合至已知癌基因。另有报道称,1例患者在接受 cilta-cel 治疗5个月后出现CAR+ T细胞淋巴瘤,其使用慢病毒载体,CAR转基因主要插入 PBX2 的3′非翻译区;该患者的白细胞单采产物中存在既存具有恶性潜能的克隆性 T 细胞群,且在 CAR-T 细胞生产前该克隆群中存在 TET2 和 JAK3 突变。在宾夕法尼亚大学接受 FDA 批准的 CAR-T 细胞产品治疗的449例患者中,1例患者在接受 axi-cel 后发生非小细胞肺癌和 T 细胞淋巴瘤;T细胞淋巴瘤不表达 CAR 转基因,但表达意义不确定的 JAK3 变体。
总体而言,在美国已输注超过27000次 FDA 批准的 CAR-T 细胞产品,CAR-T细胞源性 T 细胞恶性肿瘤极为罕见,其发病机制尚未完全了解。部分接受 CAR-T 细胞治疗的患者可能具有发生恶性肿瘤的潜在、既存遗传倾向。事实上,B细胞恶性肿瘤患者容易发生 T 细胞恶性肿瘤,反之亦然;因此,部分第二 T细胞恶性肿瘤病例可能更多地与患者的潜在遗传倾向相关,而与CAR-T细胞本身无关。
在考虑治疗选择和咨询患者时,必须权衡第二恶性肿瘤风险与成功治疗B细胞或浆细胞恶性肿瘤的潜在获益,以及与该恶性肿瘤其他化疗的风险(包括既往接受的化疗线数以及CAR-T细胞治疗的潜在替代方案)。值得注意的是,既往接受过自体HSCT的淋巴瘤患者发生治疗后第二恶性肿瘤的基线风险较高,估计所有继发性恶性肿瘤的10年累积风险为20%,继发性髓系恶性肿瘤的10年累积风险为4-6.8%。在大多数患者中,CAR-T细胞治疗的潜在获益超过第二恶性肿瘤的风险。
设计毒性更低的CAR-T细胞
降低CRS、ICANS和其他炎性毒性
尽管毒性预防和缓解策略使CAR-T细胞疗法更可行且患者更容易获得,但开发毒性更低的CAR-T细胞疗法仍是最终目标,已经提出了一系列改善CAR-T细胞疗法安全性的策略(部分策略见图2)。
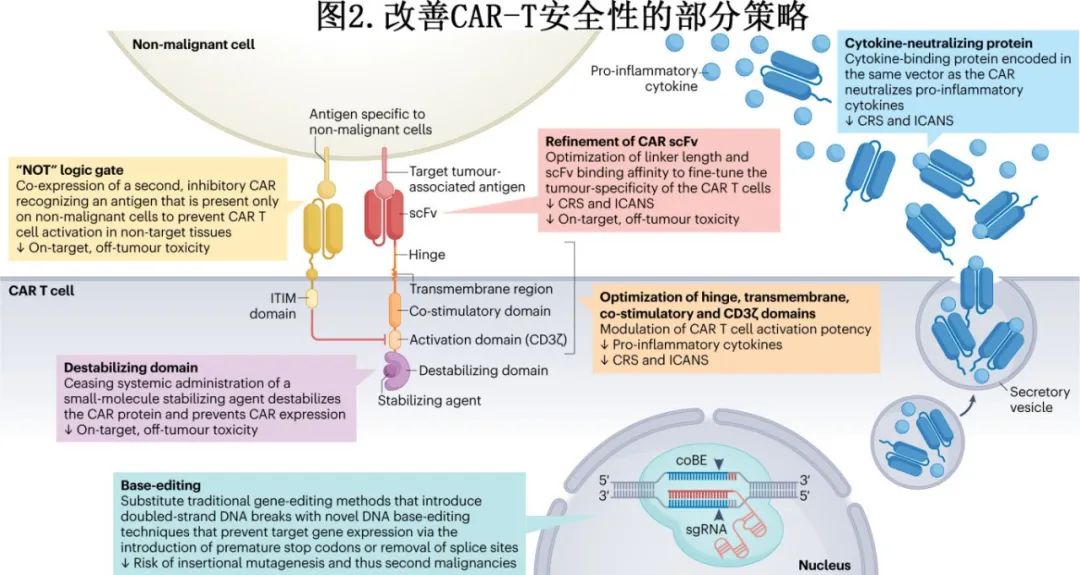
例如,已探索修饰CAR各部分以预防毒性。AUTO1是一种CD19 CAR-T细胞产品,使用单链可变片段(scFv),由于快速解离,抗原亲和力较低,在20例B-ALL患者中未导致≥3级CRS。尽管CD28共刺激结构域通常认为是CAR-T细胞产品axi-cel和brexu-cel产生严重毒性的关键驱动因子,但对其他CAR结构域的修饰降低了含CD28共刺激结构域产品的毒性。例如,与CD28铰链、跨膜和共刺激结构域(axi-cel和brexu-cel)的CAR相比,具有CD28共刺激结构域但CD8α铰链和跨膜结构域的CD19 CAR似乎具有更低的峰值血清细胞因子水平和更低的重度神经毒性发生率。I期试验还表明,在CD28共刺激和CD3ζ衍生的T细胞活化结构域之间结合细胞内toll样受体2衍生结构域可导致IFNγ产生减少,CRS和ICANS的发生率降低。在一项针对19(T2)28z1xx CAR-T细胞的LBCL患者的试验中,将CD3ζ结构域免疫受体酪氨酸激活基元的数量从3个减少到2个也导致CRS和ICANS的低发生率。
在载体中加入额外的安全成分和改变细胞培养过程可改变CAR表达细胞的炎性毒性谱。自杀基因和可用药安全开关作为毒性管理的最后手段,在消除患者体内大多数CAR-T细胞方面有一定功效;但这种CAR-T细胞消除策略的临床数据非常有限,因此它们在毒性管理中的总体有效性尚不清楚。此外,药物调节平台(drug-regulatable platforms),如CAR蛋白胞内羧基端结合的药物控制稳定或不稳定结构域,可操纵CAR的细胞表面表达(图2),提供可逆抑制并随后恢复CAR-T细胞活性的潜力。CAR-T细胞也可设计成自主分泌蛋白质来中和炎症细胞因子,如IL-1和IL-6(图2);这些细胞因子中和蛋白的序列可以利用核糖体跳变结构域整合到与CAR相同的载体中。在制备过程中,在培养中选择和促进具有较少分化表型的T细胞,从而产生更有效的CAR-T细胞,也可能导致更可控的安全性。
减轻对表达靶抗原的非恶性组织的损伤和插入性突变
编辑CAR-T细胞以避免对表达靶抗原的非恶性组织造成损伤,这是一个创新研究领域(图2)。低亲和力CAR-T细胞正在评估在靶抗原表达水平较低的非恶性组织(相对于高抗原表达的恶性组织)中预防CAR-T细胞细胞毒的潜力。逻辑门控技术也可阻止非恶性组织的靶向,包括“NOT”门控,通过结合次级抑制性CAR来阻止CAR-T细胞在遇到非恶性细胞(而非癌细胞上)的特定抗原时激活。类似地,CAR-T细胞可设计为同时表达CAR和嵌合趋化因子受体(CCR)作为第二种肿瘤相关抗原,因此CAR和CCR必须同时与其靶抗原结合才能使CAR-T细胞活化,从而增加其肿瘤特异性。
目前正在开发多种方法,以减少对基因编辑技术的需求,从而降低插入性突变的风险,特别是在异基因供者来源的CAR-T细胞的情况下(为了避免移植物排斥和移植物抗宿主病,需要进行多次基因编辑步骤)。碱基编辑技术可能是目前使用的基因编辑技术(比如传统的CRISPR和TALEN,会产生DNA断裂)的可行替代品。对于正在开发用于治疗T细胞白血病和淋巴瘤的CD7 CAR-T细胞,通过从CAR介导的细胞内CD7隔离或表位掩蔽的培养物中自然选择CAR-T细胞,可使第二步基因编辑敲低CAR-T细胞上的CD7表达,以避免自相残杀。
总结
随着成千上万的患者接受CAR-T细胞治疗,急性毒性(如CRS和ICANS)的管理得到显著改善,许多专家能够自如地管理这些毒性。然而,对一些以前不太为人所知的CAR-T细胞相关毒性(如ICAHT、IEC-HS和运动障碍)的病理生理学的理解仍然有限,治疗策略也在不断发展。在未来几年,继续对毒性干预方法进行前瞻性评估和更新治疗原则将至关重要。此外,开发新型CAR-T细胞结构和具有更有利毒性特征的产品将使患者更容易获得这些突破性疗法。
参考文献
Brudno JN, Kochenderfer JN. Current understanding and management of CAR T cell-associated toxicities.Nat Rev Clin Oncol . 2024 Jul;21(7):501-521. doi: 10.1038/s41571-024-00903-0.

